The Development and Validation of a Novel “Dual Cocktail” Probe for Cytochrome P450s and Transporter Functions to Evaluate Pharmacokinetic Drug-Drug and Herb-Drug Interactions


Abstract
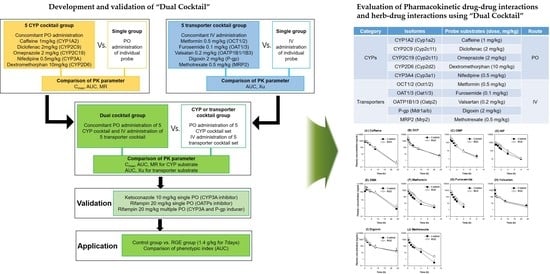
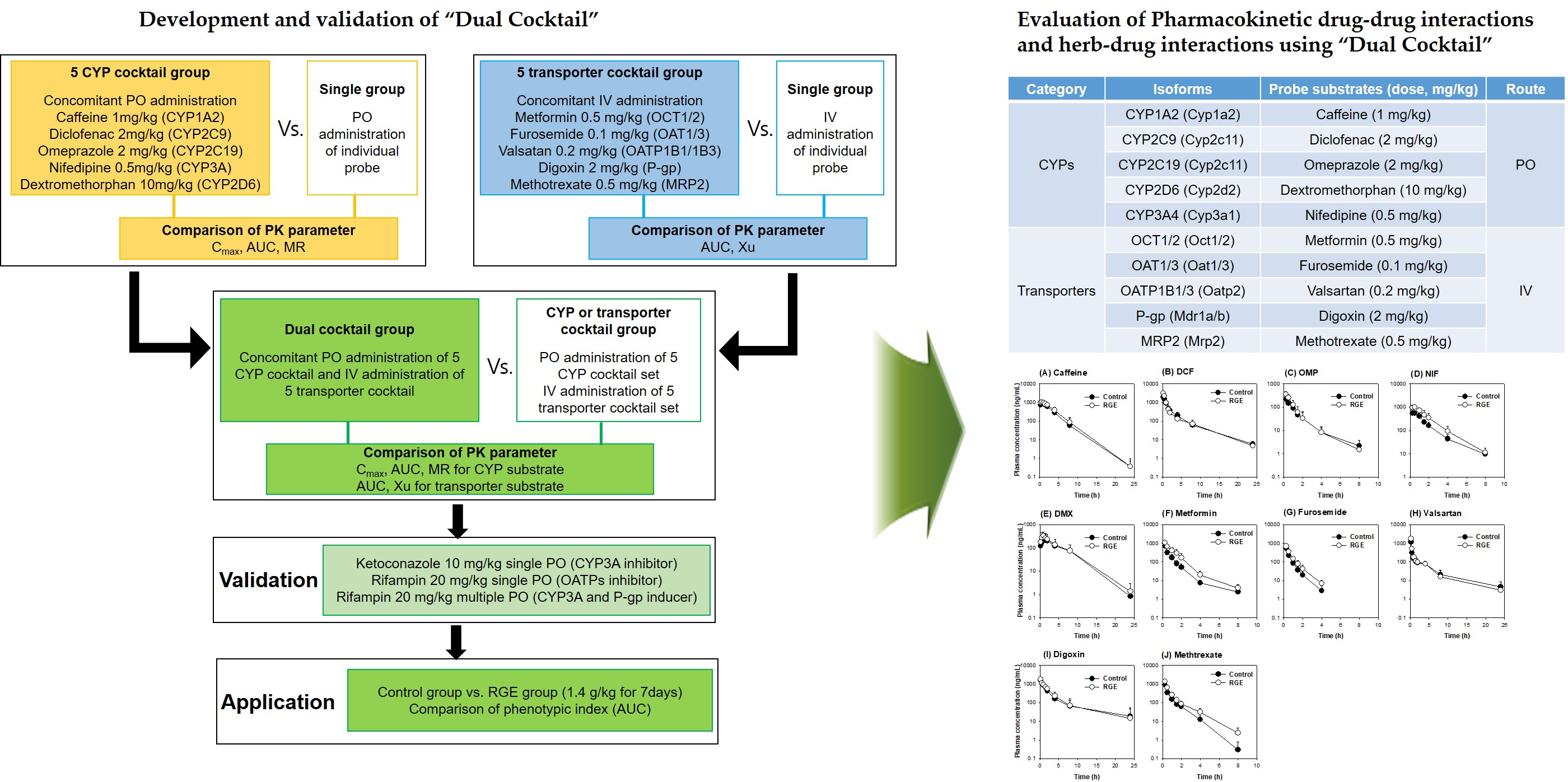
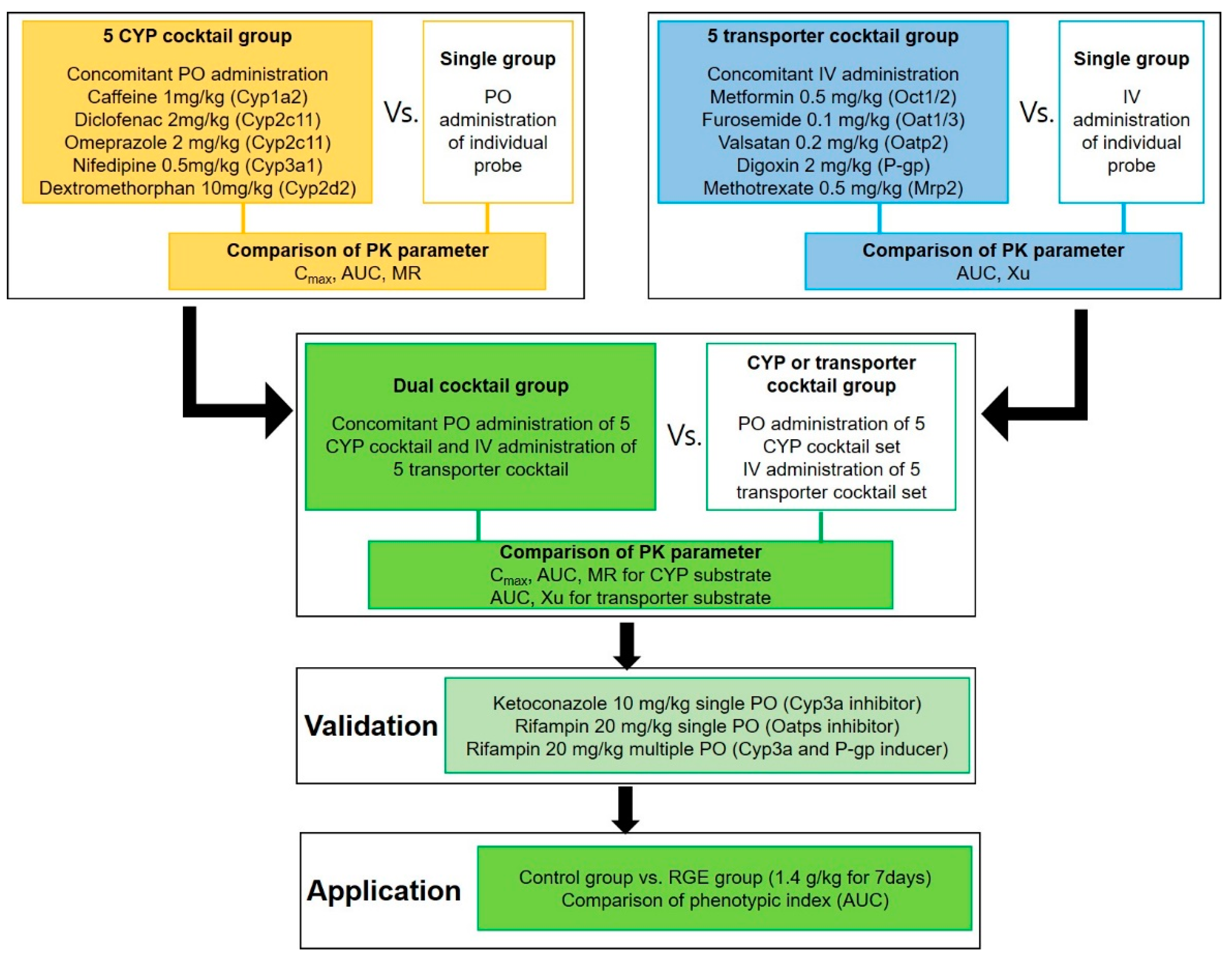
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals and Ethical Approval
2.3. Pharmacokinetic Study
2.4. LC-MS/MS Analysis of Probe Substrate
2.5. Data Analysis
3. Results
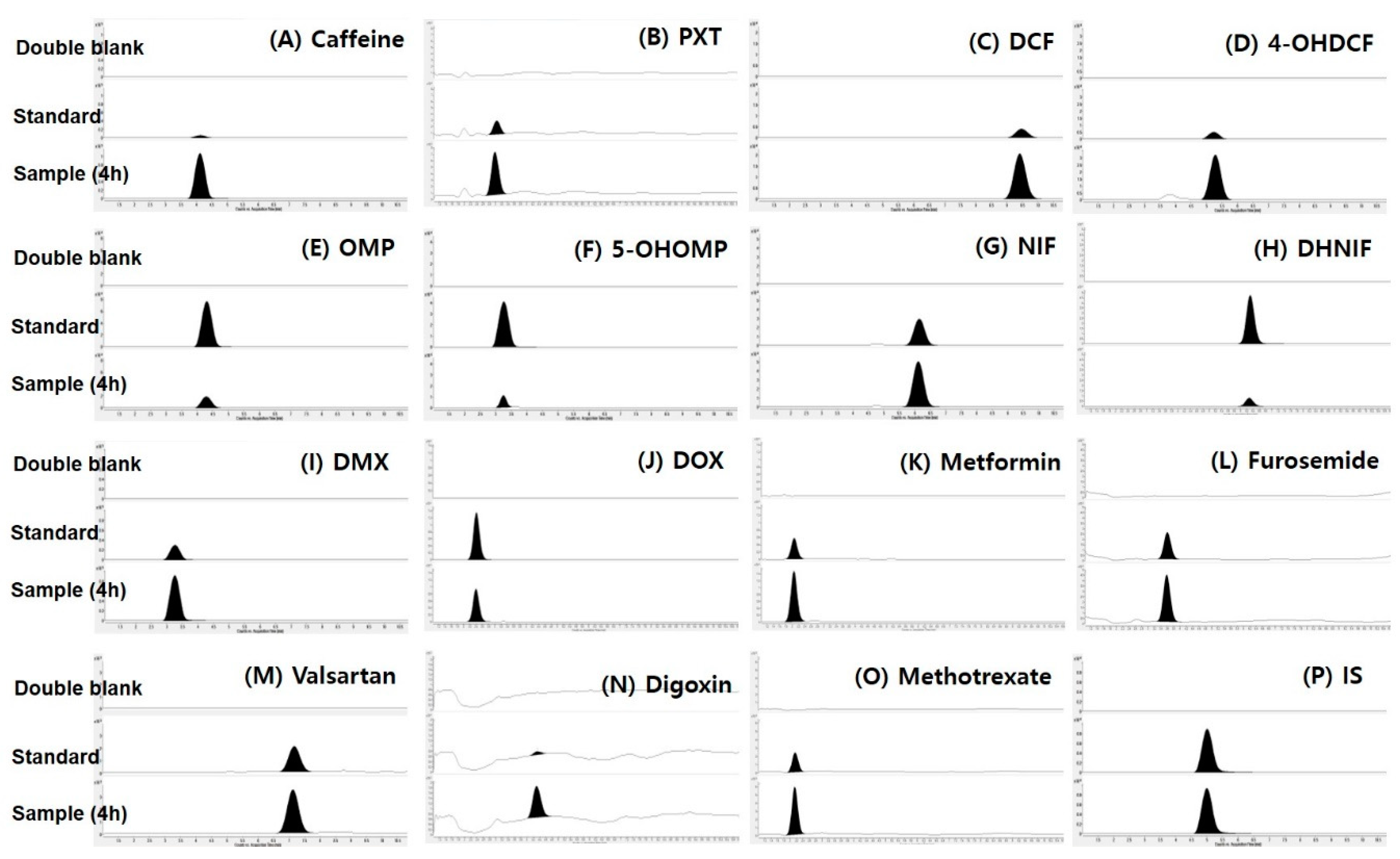
3.1. Simultaneous Determination of Selected Probe Substrate for CYP Enzymes and Transporters
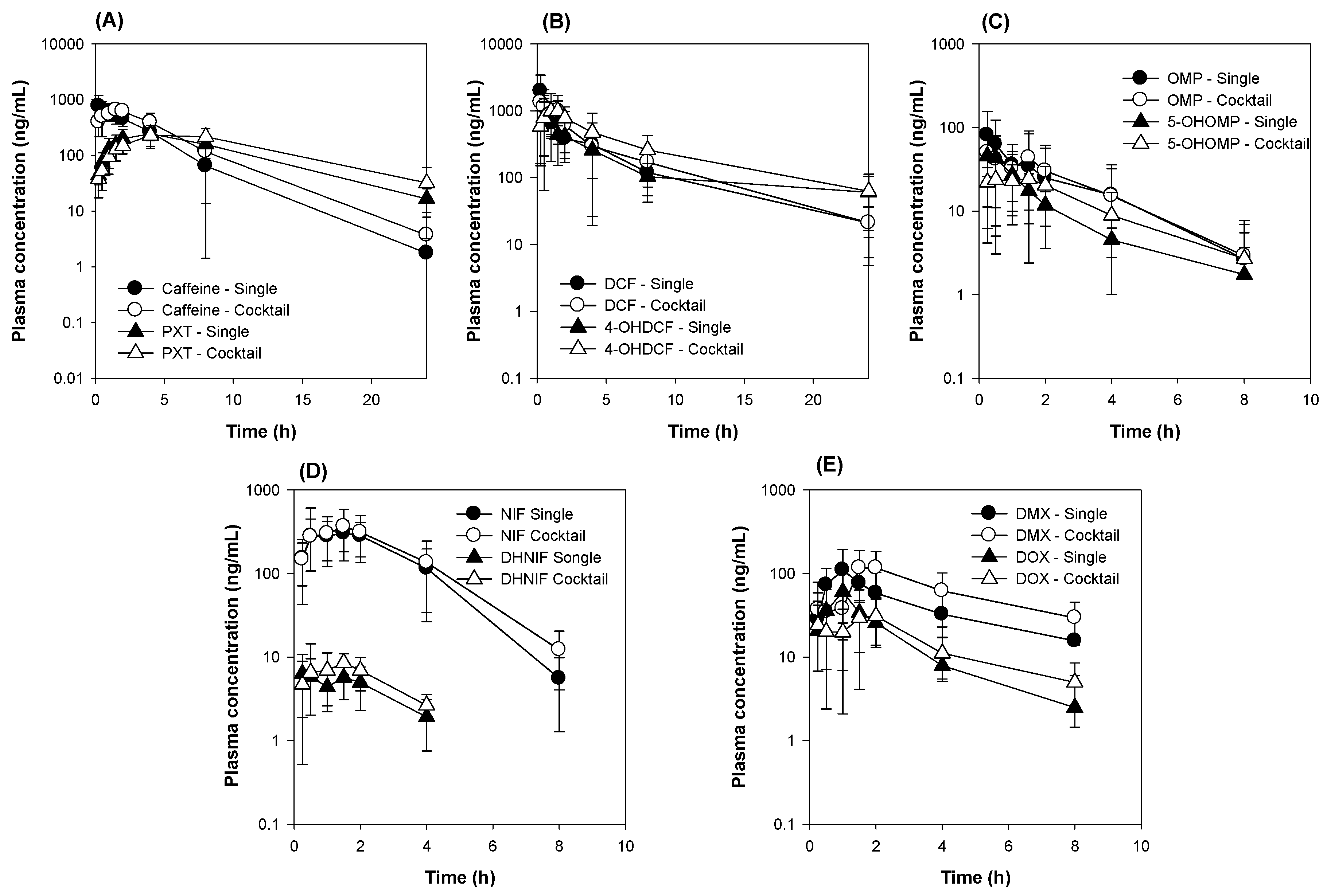
3.2. Comparison of Five CYP Probe Substrate Simultaneously versus Individually Administered
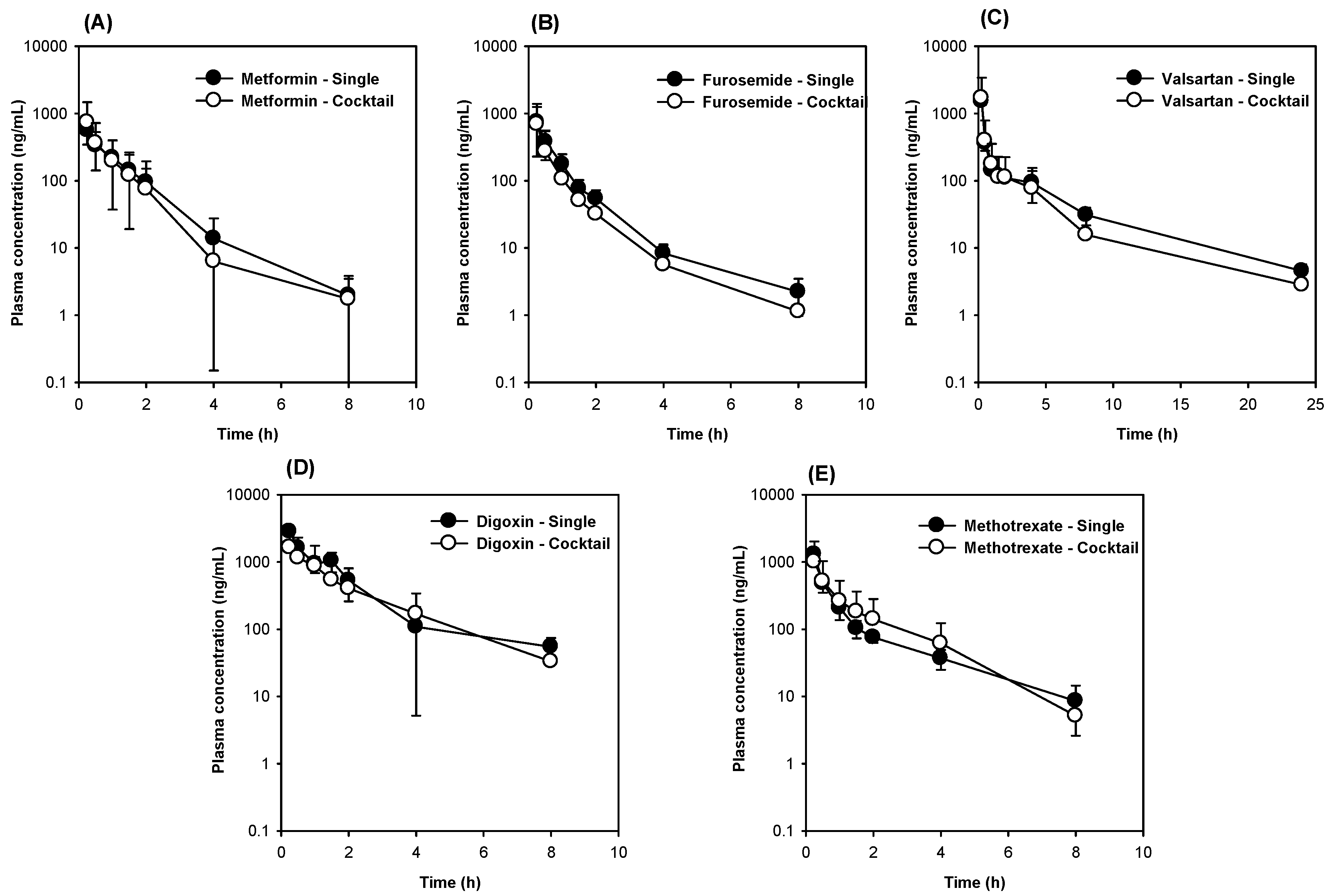
3.3. The Comparison of Five Transporter Probe Substrates Administered Simultaneously versus Individually
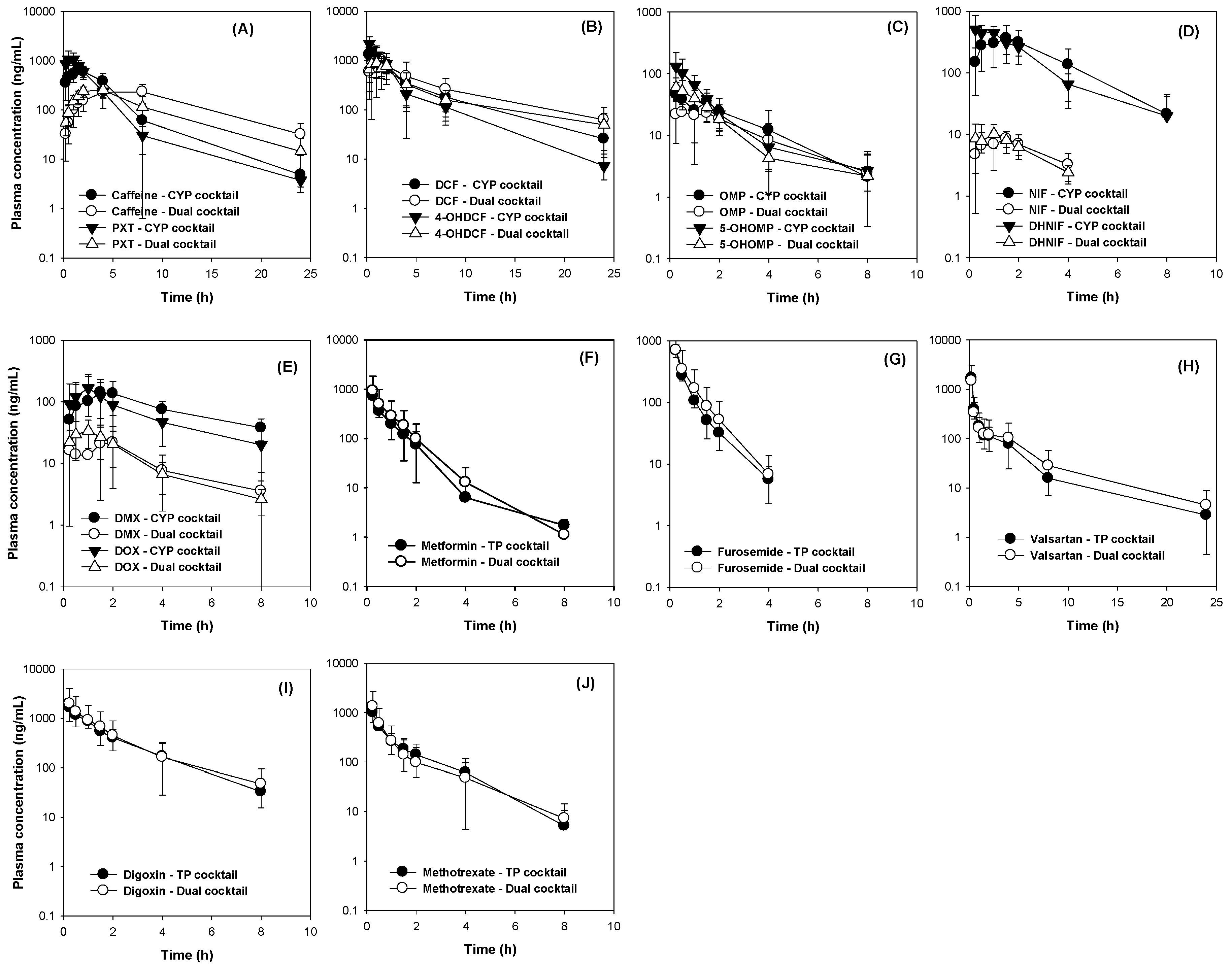
3.4. The Comparison of Pharmacokinetics Parameters Following Administration of Dual Cocktail vs. Five Transporter or Five CYP Cocktail
3.5. Dual Cocktail Validation by Evaluating the Effects of Known CYP or Transporter Modulators on the Pharmacokinetic Parameters of Probe Substrates
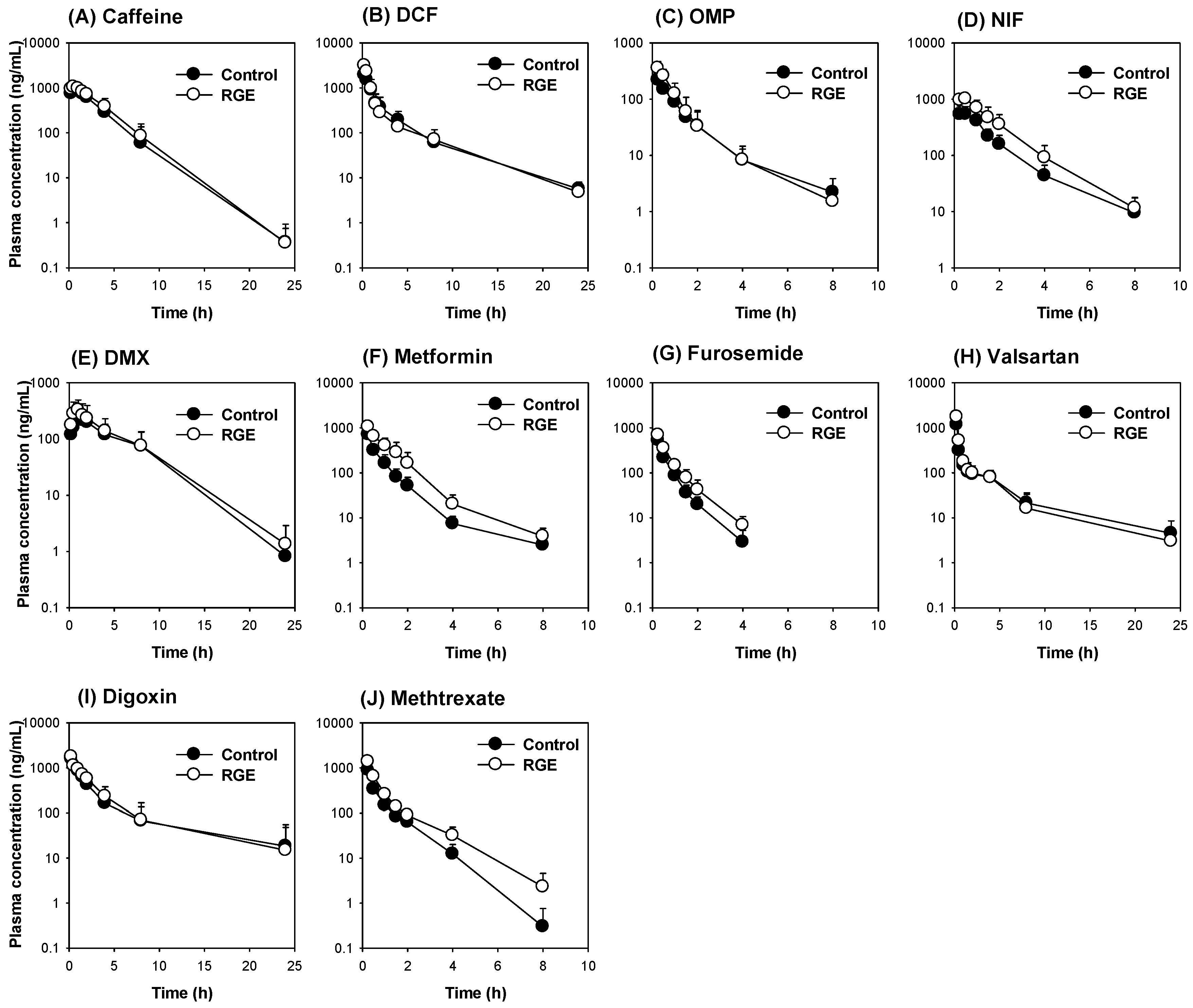
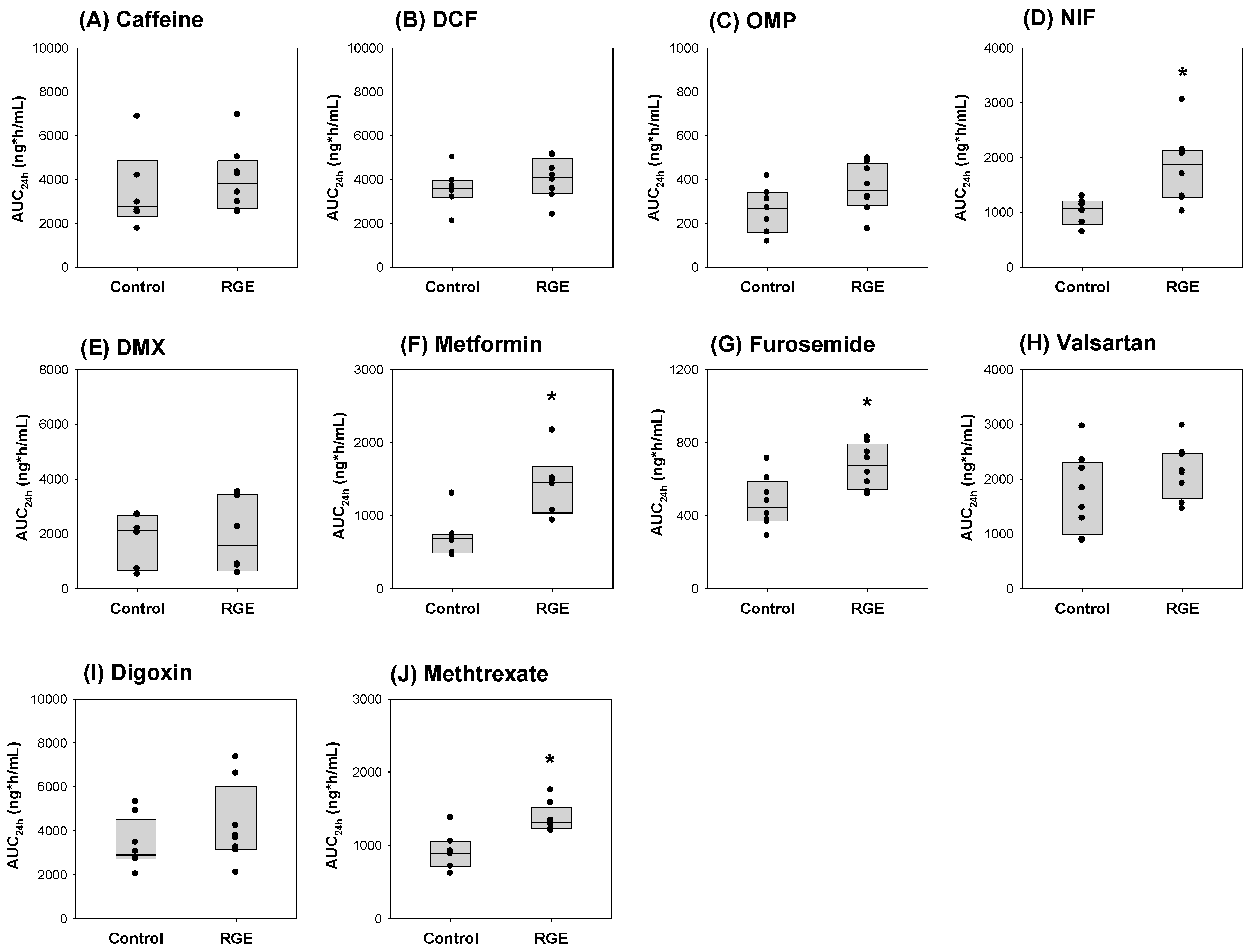
3.6. The Use of the Dual Cocktail to Assess the Pharmacokinetic Herb-Drug Interaction of RGE Treatment
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Choi, M.K.; Song, I.S. Interactions of ginseng with therapeutic drugs. Arch. Pharmacal Res. 2019, 42, 862–878. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ma, J.; Ye, Y.; Lin, G. Transporter modulation by Chinese herbal medicines and its mediated pharmacokinetic herb-drug interactions. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1026, 236–253. [Google Scholar] [CrossRef] [PubMed]
- Adiwidjaja, J.; Boddy, A.V.; McLachlan, A.J. Physiologically based pharmacokinetic modelling of hyperforin to predict drug interactions with St John’s wort. Clin. Pharmacokinet. 2019, 58, 911–926. [Google Scholar] [CrossRef]
- Brantley, S.; Graf, T.N.; Oberlies, N.H.; Paine, M.F. Physiologically-based pharmacokinetic interaction model framework for quantitative prediction of herb-drug interactions. Drug Metab. Rev. 2012, 44, 99–100. [Google Scholar]
- Bosilkovska, M.; Samer, C.F.; Deglon, J.; Rebsamen, M.; Staub, C.; Dayer, P.; Walder, B.; Desmeules, J.A.; Daali, Y. Geneva cocktail for Cytochrome P450 and P-glycoprotein activity assessment using dried blood spots. Clin. Pharmacol. Ther. 2014, 96, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Derungs, A.; Donzelli, M.; Berger, B.; Noppen, C.; Krahenbuhl, S.; Haschke, M. Effects of Cytochrome P450 inhibition and induction on the phenotyping metrics of the Basel Cocktail: A randomized crossover study. Clin. Pharmacokinet. 2016, 55, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.Y.; Song, I.S.; Sunwoo, Y.E.; Shon, J.H.; Liu, K.H.; Cha, I.J.; Shin, J.G. Development of the “Inje cocktail” for high-throughput evaluation of five human cytochrome p450 Isoforms in vivo. Clin. Pharmacol. Ther. 2007, 82, 531–540. [Google Scholar] [CrossRef]
- Uchida, S.; Tanaka, S.; Namiki, N. Simultaneous and comprehensive in vivo analysis of cytochrome P450 activity by using a cocktail approach in rats. Biopharm. Drug Dispos. 2014, 35, 228–236. [Google Scholar] [CrossRef]
- Videau, O.; Delaforge, M.; Levi, M.; Thevenot, E.; Gal, O.; Becquemont, L.; Beaune, P.; Benech, H. Biochemical and analytical development of the CIME cocktail for drug fate assessment in humans. Rapid Commun. Mass Spectrom. 2010, 24, 2407–2419. [Google Scholar] [CrossRef]
- Videau, O.; Pitarque, S.; Troncale, S.; Hery, P.; Thevenot, E.; Delaforge, M.; Benech, H. Can a cocktail designed for phenotyping pharmacokinetics and metabolism enzymes in human be used efficiently in rat? Xenobiotica 2012, 42, 349–354. [Google Scholar] [CrossRef]
- International Transporter Consortium; Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Sotaniemi, E.A.; Haapakoski, E.; Rautio, A. Ginseng therapy in non-insulin-dependent diabetic patients. Diabetes Care 1995, 18, 1373–1375. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kwon, M.; Choi, M.K.; Song, I.S. Effects of red ginseng extract on the pharmacokinetics and elimination of methotrexate via Mrp2 regulation. Molecules 2018, 23, 2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA. In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions (accessed on 30 September 2020).
- EMA. Guideline on the investigation of drug interactions. React. Wkly. 2012. [Google Scholar] [CrossRef]
- Liska, D.J. The detoxification enzyme systems. Altern. Med. Rev. 1998, 3, 187–198. [Google Scholar]
- Choi, J.S.; Choi, I.; Choi, D.H. Effects of pioglitazone on the pharmacokinetics of nifedipine and its main metabolite, dehydronifedipine, in rats. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 231–238. [Google Scholar] [CrossRef]
- Choi, Y.H.; Lee, M.G. Pharmacokinetic and pharmacodynamic interaction between nifedipine and metformin in rats: Competitive inhibition for metabolism of nifedipine and metformin by each other via CYP isozymes. Xenobiotica 2012, 42, 483–495. [Google Scholar] [CrossRef]
- Chovan, J.P.; Ring, S.C.; Yu, E.; Baldino, J.P. Cytochrome P450 probe substrate metabolism kinetics in Sprague Dawley rats. Xenobiotica 2007, 37, 459–473. [Google Scholar] [CrossRef]
- Yamashiro, W.; Maeda, K.; Hirouchi, M.; Adachi, Y.; Hu, Z.; Sugiyama, Y. Involvement of transporters in the hepatic uptake and biliary excretion of valsartan, a selective antagonist of the angiotensin II AT1-receptor, in humans. Drug Metab. Dispos. 2006, 34, 1247–1254. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Ma, J.; Li, T.; Xiao, Y.; Zhang, N.; Liu, J.; Gao, Y.; Shao, J.; Jia, L. Global deregulation of ginseng products may be a safety hazard to warfarin takers: Solid evidence of ginseng-warfarin interaction. Sci. Rep. 2017, 7, 5813. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.X.; Ritchie, D.M.; Cousineau, M.; Danser, E.; Dewire, R.; Floden, J. Altered oral bioavailability and pharmacokinetics of P-glycoprotein substrates by coadministration of biochanin A. J. Pharmacol. Sci. 2006, 95, 1984–1993. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Han, K.S.; Lee, M.G. Gastrointestinal first-pass effect of furosemide in rats. J. Pharm. Pharmacol. 2000, 52, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Kim, S.G.; Lee, M.G. Dose-independent pharmacokinetics of metformin in rats: Hepatic and gastrointestinal first-pass effects. J. Pharmacol. Sci. 2006, 95, 2543–2552. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kang, W.S.; Piao, J.; Yoon, I.S.; Kim, D.D.; Cho, H.J. Soluplus (R)/TPGS-based solid dispersions prepared by hot-melt extrusion equipped with twin-screw systems for enhancing oral bioavailability of valsartan. Drug Des. Dev. Ther. 2015, 9, 2745–2756. [Google Scholar]
- Chen, C.P.; Scott, D.; Hanson, E.; Franco, J.; Berryman, E.; Volberg, M.; Liu, X.R. Impact of mrp2 on the biliary excretion and intestinal absorption of furosemide, probenecid, and methotrexate using Eisai hyperbilirubinemic rats. Pharm. Res. 2003, 20, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kim, J.H. A review on the medicinal potentials of ginseng and ginsenosides on cardiovascular diseases. J. Ginseng Res. 2014, 38, 161–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ru, W.; Wang, D.; Xu, Y.; He, X.; Sun, Y.E.; Qian, L.; Zhou, X.; Qin, Y. Chemical constituents and bioactivities of Panax ginseng (C. A. Mey.). Drug Discov. Ther. 2015, 9, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Gurley, B.J.; Gardner, S.F.; Hubbard, M.A.; Williams, D.K.; Gentry, W.B.; Cui, Y.; Ang, C.Y. Clinical assessment of effects of botanical supplementation on cytochrome P450 phenotypes in the elderly: St John’s wort, garlic oil, Panax ginseng and Ginkgo biloba. Drugs Aging 2005, 22, 525–539. [Google Scholar] [CrossRef]
- Gurley, B.J.; Gardner, S.F.; Hubbard, M.A.; Williams, D.K.; Gentry, W.B.; Cui, Y.; Ang, C.Y. Cytochrome P450 phenotypic ratios for predicting herb-drug interactions in humans. Clin. Pharmacol. Ther. 2002, 72, 276–287. [Google Scholar] [CrossRef]
- Kim, D.S.; Kim, Y.; Jeon, J.Y.; Kim, M.G. Effect of red ginseng on cytochrome P450 and P-glycoprotein activities in healthy volunteers. J. Ginseng Res. 2016, 40, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Seong, S.J.; Kang, W.Y.; Heo, J.K.; Jo, J.; Choi, W.G.; Liu, K.H.; Lee, S.; Choi, M.K.; Han, Y.H.; Lee, H.S.; et al. A comprehensive in vivo and in vitro assessment of the drug interaction potential of red ginseng. Clin. Ther. 2018, 40, 1322–1337. [Google Scholar] [CrossRef] [PubMed]
- Malati, C.Y.; Robertson, S.M.; Hunt, J.D.; Chairez, C.; Alfaro, R.M.; Kovacs, J.A.; Penzak, S.R. Influence of panax ginseng on cytochrome P450 (CYP)3A and P-glycoprotein (P-gp) activity in healthy participants. J. Clin. Pharmacol. 2012, 52, 932–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Nam, W.; Kim, S.H.; Jang, H.R.; Lee, M.K.; Kim, T.W.; Lee, S. Modulatory effects of Korean red ginseng extract (Panax ginseng C.A. Meyer) on cytochrome P450 after oral administration to mice for 14 days. J. Life Sci. 2012, 22, 991–998. [Google Scholar] [CrossRef] [Green Version]
- Bogacz, A.; Karasiewicz, M.; Dziekan, K.; Procyk, D.; Górska-Paukszta, M.; Kowalska, A.; Mikolajczak, P.L.; Ożarowski, M.; Czerny, B. Impact of Panax ginseng and Ginkgo biloba extracts on expression level of transcriptional factors and xenobiotic-metabolizing cytochrome P450 enzymes. Herba Pol. 2016, 62, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Hong, B.N.; Ji, M.G.; Kang, T.H. The efficacy of red ginseng in type 1 and type 2 diabetes in animals. Evid. Based Complement. Altern. Med. 2013, 2013, 593181. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.J.; Kim, N.; Lee, K.; Hee Sonn, C.; Eun Lee, J.; Tae Kim, S.; Ho Baeg, I.; Lee, K.M. Korean red ginseng (Panax ginseng) ameliorates type 1 diabetes and restores immune cell compartments. J. Ethnopharmacol. 2012, 144, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Hyun, H.B.; Lee, W.S.; Go, S.I.; Nagappan, A.; Park, C.; Han, M.H.; Hong, S.H.; Kim, G.; Kim, G.Y.; Cheong, J.; et al. The flavonoid morin from Moraceae induces apoptosis by modulation of Bcl-2 family members and Fas receptor in HCT 116 cells. Int. J. Oncol. 2015, 46, 2670–2678. [Google Scholar] [CrossRef] [Green Version]
- Nam, S.J.; Han, Y.J.; Lee, W.; Kang, B.; Choi, M.K.; Han, Y.H.; Song, I.S. Effect of red ginseng extract on the pharmacokinetics and efficacy of metformin in streptozotocin-induced diabetic rats. Pharmaceutics 2018, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, Z.; Huang, Y.; O’Barr, S.A.; Wong, R.A.; Yeung, S.; Chow, M.S. Ginseng and anticancer drug combination to improve cancer chemotherapy: A critical review. Evid. Based Complement. Alternat. Med. 2014, 2014, 168940. [Google Scholar] [CrossRef]
- Endale, M.; Im, E.J.; Lee, J.Y.; Kim, S.D.; Yayeh, T.; Song, Y.B.; Kwak, Y.S.; Kim, C.; Kim, S.H.; Roh, S.S.; et al. Korean red ginseng saponin fraction rich in ginsenoside-Rb1, Rc and Rb2 attenuates the severity of mouse collagen-induced arthritis. Mediat. Inflamm. 2014, 2014, 748964. [Google Scholar] [CrossRef]
- Zheng, H.X.; Huang, Y.; Frassetto, L.A.; Benet, L.Z. Elucidating rifampin’s inducing and inhibiting effects on glyburide pharmacokinetics and blood glucose in healthy volunteers: Unmasking the differential effects of enzyme induction and transporter inhibition for a drug and its primary metabolite. Clin. Pharmacol. Ther. 2009, 85, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.K.; Kim, H.J.; Lee, G.H.; Ohk, B.; Lee, S.; Song, K.S.; Song, I.S.; Liu, K.H.; Yoon, Y.R. Simultaneous determination of five Cytochrome P450 probe substrates and their metabolites and organic anion transporting polypeptide probe substrate in human plasma using liquid chromatography-tandem mass spectrometry. Pharmaceutics 2018, 10, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Lee, S.; Jeon, J.H.; Kim, H.; Choi, M.K.; Song, I.S. Enhanced intestinal permeability and plasma concentration of metformin in rats by the repeated administration of red ginseng extract. Pharmaceutics 2019, 11, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, J.H.; Lee, S.; Lee, W.; Jin, S.; Kwon, M.; Shin, C.H.; Choi, M.K.; Song, I.S. Herb-drug interaction of red ginseng extract and ginsenoside Rc with valsartan in rats. Molecules 2020, 25, 622. [Google Scholar] [CrossRef] [Green Version]
- Frommherz, L.; Kohler, H.; Brinkmann, B.; Lehr, M.; Beike, J. LC-MS assay for quantitative determination of cardio glycoside in human blood samples. Int. J. Leg. Med. 2008, 122, 109–114. [Google Scholar] [CrossRef]
- Keane, S.; Wallace, G.; Munday, C.; Wright, M. Sensitive and robust LC-MS/MS analysis of digoxin in human plasma through optimization of in-source adduct formation. Bioanalysis 2018, 10, 1401–1411. [Google Scholar] [CrossRef]
- Kim, S.; Choi, W.G.; Kwon, M.; Lee, S.; Cho, Y.Y.; Lee, J.Y.; Kang, H.C.; Song, I.S.; Lee, H.S. In vitro inhibitory effects of APINACA on human major Cytochrome P450, UDP-Glucuronosyltransferase enzymes, and drug transporters. Molecules 2019, 24, 3000. [Google Scholar] [CrossRef] [Green Version]
- Song, I.S.; Kong, T.Y.; Jeong, H.U.; Kim, E.N.; Kwon, S.S.; Kang, H.E.; Choi, S.Z.; Son, M.; Lee, H.S. Evaluation of the transporter-mediated herb-drug interaction potential of DA-9801, a standardized dioscorea extract for diabetic neuropathy, in human in vitro and rat in vivo. BMC Complement. Altern. Med. 2014, 14, e251. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, D. New US FDA draft guidance on bioanalytical method validation versus current FDA and EMA guidelines: Chromatographic methods and ISR. Bioanalysis 2014, 6, 13–19. [Google Scholar] [CrossRef]
- Wang, J.S.; Wen, X.; Backman, J.T.; Taavitsainen, P.; Neuvonen, P.J.; Kivisto, K.T. Midazolam alpha-hydroxylation by human liver microsomes in vitro: Inhibition by calcium channel blockers, itraconazole and ketoconazole. Pharmacol. Toxicol. 1999, 85, 157–161. [Google Scholar] [CrossRef]
- Lee, K.S.; Park, J.H.; Lee, H.J.; Rhie, S. Preclinical study of a potent P-glycoprotein and cytochrome P450 enzyme inducer rifampicin changing pharmacokinetic parameters of risperidone and its metabolite, 9-hydroxyrisperidone, using a rat model. J. Pharm. Investig. 2012, 42, 345–351. [Google Scholar] [CrossRef]
- Yigitaslan, S.; Erol, K.; Cengelli, C. The effect of P-glycoprotein Inhibition and activation on the absorption and serum levels of cyclosporine and tacrolimus in rats. Adv. Clin. Exp. Med. 2016, 25, 237–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.H.; Ly, J.; Plise, E.; Zhang, X.; Messick, K.; Wright, M.; Cheong, J. Differential effects of rifampin and ketoconazole on the blood and liver concentration of atorvastatin in wild-type and Cyp3a and Oatp1a/b knockout mice. Drug. Metab. Dispos. 2014, 42, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, C.; Yin, O.Q.; Smith, T.; Sethuraman, V.; Grouss, K.; Galitz, L.; Harrell, R.; Schran, H. Effects of rifampin and ketoconazole on the pharmacokinetics of nilotinib in healthy participants. J. Clin. Pharmacol. 2011, 51, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Cho, H.Y.; Lee, Y.B. Effect of cimetidine and phenobarbital on metabolite kinetics of omeprazole in rats. Arch. Pharmacal Res. 2005, 28, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Takashima, T.; Murase, S.; Iwasaki, K.; Shimada, K. Evaluation of dextromethorphan metabolism using hepatocytes from CYP2D6 poor and extensive metabolizers. Drug Metab. Pharmacokinet. 2005, 20, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Masuda, M.; I’Izuka, Y.; Yamazaki, M.; Nishigaki, R.; Kato, Y.; Ni’inuma, K.; Suzuki, H.; Sugiyama, Y. Methotrexate is excreted into the bile by canalicular multispecific organic anion transporter in rats. Cancer Res. 1997, 57, 3506–3510. [Google Scholar]
- Poirier, A.; Cascais, A.C.; Funk, C.; Lave, T. Prediction of pharmacokinetic profile of valsartan in human based on in vitro uptake transport data. J. Pharmacokinet. Pharmacodyn. 2009, 36, 585–611. [Google Scholar] [CrossRef]
- Sager, J.E.; Yu, J.; Ragueneau-Majlessi, I.; Isoherranen, N. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: A systematic review of published models, applications, and model verification. Drug Metab. Dispos. 2015, 43, 1823–1837. [Google Scholar] [CrossRef]
- Brantley, S.J.; Argikar, A.A.; Lin, Y.S.; Nagar, S.; Paine, M.F. Herb-drug interactions: Challenges and opportunities for improved predictions. Drug Metab. Dispos. 2014, 42, 301–317. [Google Scholar] [CrossRef] [Green Version]
- Lewis, D.F. Homology modelling of human cytochromes P450 involved in xenobiotic metabolism and rationalization of substrate selectivity. Exp. Toxicol. Pathol. 1999, 51, 369–374. [Google Scholar] [CrossRef]
- Sakai, C.; Iwano, S.; Yamazaki, Y. Species Differences in the Pharmacokinetic Parameters of Cytochrome P450 Probe Substrates between Experimental Animals, such as Mice, Rats, Dogs, Monkeys, and Microminipigs, and Humans. J. Drug Metab. Toxicol. 2014, 5, 1000173. [Google Scholar]
- Martignoni, M.; Groothuis, G.M.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.Y.; Bleasby, K.; Evers, R. Species differences in drug transporters and implications for translating preclinical findings to humans. Expert Opin. Drug Metab. Toxicol. 2013, 9, 237–252. [Google Scholar] [CrossRef]
- Choi, M.K.; Song, I.S. Organic Cation Transporters and their Pharmacokinetic and Pharmacodynamic Consequences. Drug Metab. Pharmacokinet. 2008, 23, 243–253. [Google Scholar] [CrossRef]
- Feng, B.; Mills, J.B.; Davidson, R.E.; Mireles, R.J.; Janiszewski, J.S.; Troutman, M.D.; de Morais, S.M. In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab. Dispos. 2008, 36, 268–275. [Google Scholar] [CrossRef]
- Svanstrom, H.; Lund, M.; Melbye, M.; Pasternak, B. Concomitant use of low-dose methotrexate and NSAIDs and the risk of serious adverse events among patients with rheumatoid arthritis. Pharmacoepidemiol. Drug Saf. 2018, 27, 885–893. [Google Scholar] [CrossRef]
- Ni, Y.; Duan, Z.L.; Zhou, D.D.; Liu, S.; Wan, H.D.; Gui, C.S.; Zhang, H.J. Identification of structural features for the inhibition of OAT3-mediated uptake of enalaprilat by selected drugs and flavonoids. Front. Pharmacol. 2020, 11, e802. [Google Scholar] [CrossRef]
- Kothari, N.; Ganguly, B. Potential drug—drug interactions among medications prescribed to hypertensive patients. J. Clin. Diagn. Res. 2014, 8, HC01–HC04. [Google Scholar] [CrossRef]
- Greiner, B.; Eichelbaum, M.; Fritz, P.; Kreichgauer, H.P.; Von Richter, O.; Zundler, J.; Kroemer, H.K. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J. Clin. Investig. 1999, 104, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Kimoto, E.; Vourvahis, M.; Scialis, R.J.; Eng, H.; Rodrigues, A.D.; Varma, M.V.S. Mechanistic evaluation of the complex drug-drug interactions of maraviroc: Contribution of Cytochrome P450 3A, P-glycoprotein and Organic Anion Transporting Polypeptide 1B1. Drug Metab. Dispos. 2019, 47, 493–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaruratanasirikul, S.; Sriwiriyajan, S. Effect of omeprazole on the pharmacokinetics of itraconazole. Eur. J. Clin. Pharmacol. 1998, 54, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; Hamilton, C.D.; Drew, R.H.; Sanders, L.L.; Pennick, G.J.; Perfect, J.R. A randomized comparative study to determine the effect of omeprazole on the peak serum concentration of itraconazole oral solution. J. Antimicrob. Chemother. 2003, 51, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remmel, R.P.; Amoh, K.; Abdel-Monem, M.M. The disposition and pharmacokinetics of ketoconazole in the rat. Drug Metab. Dispos. 1987, 15, 735–739. [Google Scholar]
- Segawa, K.; Nakazawa, S.; Tsukamoto, Y.; Chujoh, C.; Yamao, K.; Hase, S. Effect of omeprazole on gastric acid secretion in rat: Evaluation of dose, duration of effect, and route of administration. Gastroenterol. Jpn. 1987, 22, 413–418. [Google Scholar] [CrossRef]
- Oishi, M.; Takano, Y.; Torita, Y.; Malhotra, B.; Chiba, K. Physiological based pharmacokinetic modeling to estimate in vivo Ki of ketoconazole on renal P-gp using human drug-drug interaction study result of fesoterodine and ketoconazole. Drug Metab. Pharmacokinet. 2018, 33, 90–95. [Google Scholar] [CrossRef]
- Takeuchi, T.; Nonaka, M.; Yoshitomi, S.; Higuchi, T.; Ebihara, T.; Maeshiba, Y.; Kawase, M.; Asahi, S. Marked impact of P-glycoprotein on the absorption of TAK-427 in rats. Biopharm. Drug Dispos. 2008, 29, 311–323. [Google Scholar] [CrossRef]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ginsenosides | Content (mg/g RGE) | Total (mg/g RGE) | |
|---|---|---|---|
| Protopanaxadiol-type | Ginsenoside Rb1 | 4.7 | 14.3 |
| Ginsenoside Rb2 | 2.3 | ||
| Ginsenoside Rc | 2.5 | ||
| Ginsenoside Rd | 1.3 | ||
| Ginsenoside Rg3 | 3.5 | ||
| Protopanaxatriol-type | Ginsenoside Re | 1.3 | 4.6 |
| Ginsenoside Rg1 | 0.6 | ||
| Ginsenoside Rf | 1.1 | ||
| Ginsenoside Rh1 | 1.6 | ||
| Analyte | Mode | m/z Q1 → Q3 | Collision Energy (V) | Retention Time (min) | Linear Range (ng/mL) |
|---|---|---|---|---|---|
| Caffeine | positive | 194.9 → 138.0 | 20 | 4.09 | 1–1000 |
| Paraxanthine | positive | 180.7 → 124.3 | 10 | 2.97 | 1–1000 |
| Diclofenac | positive | 295.9 → 215.0 | 20 | 9.58 | 1–1000 |
| 4-Hydroxydiclofenac | positive | 312.2 → 230.9 | 20 | 5.25 | 1–1000 |
| Dextromethorphan | positive | 272.0 → 214.9 | 25 | 3.37 | 1–1000 |
| Dextrorphan | positive | 258.0 → 201.0 | 25 | 2.42 | 1–1000 |
| Nifedipine | positive | 347.0 → 315.0 | 5 | 6.16 | 1–1000 |
| Dehydronifedipine | positive | 245.0 → 283.9 | 30 | 6.44 | 1–1000 |
| Omeprazole | positive | 346.0 → 197.8 | 5 | 4.29 | 1–1000 |
| 5-Hydroxyomeprazole | positive | 362.2 → 213.9 | 10 | 3.26 | 1–1000 |
| Digoxin | positive | 803.4 → 283.0 | 50 | 4.35 | 5–2000 |
| Furosemide | negative | 328.9 → 284.8 | 15 | 3.62 | 1–2000 |
| Metformin | positive | 129.9 → 60.0 | 10 | 2.05 | 1–2000 |
| Methotrexate | positive | 455.1 → 308.2 | 20 | 2.23 | 1–2000 |
| Valsartan | positive | 436.1 → 291.0 | 5 | 7.22 | 1–2000 |
| Berberine | positive | 336.1 → 320.0 | 30 | 5.07 |
| Analyte | Spiked Concentration (ng/mL) | Intra-Day | Inter-Day | ||||
|---|---|---|---|---|---|---|---|
| Measured (ng/mL) | Precision (%) | Accuracy (%) | Measured (ng/mL) | Precision (%) | Accuracy (%) | ||
| Caffeine | 5 | 4.65 ± 0.41 | 8.78 | 93.07 | 4.78 ± 0.41 | 8.66 | 95.51 |
| 50 | 46.42 ± 1.94 | 4.18 | 92.84 | 48.31 ± 5.70 | 11.81 | 96.63 | |
| 250 | 253.58 ± 11.79 | 4.65 | 101.43 | 273.24 ± 24.11 | 8.83 | 109.30 | |
| Paraxanthine | 5 | 5.17 ± 0.40 | 7.76 | 103.44 | 5.17 ± 0.40 | 7.76 | 103.44 |
| 50 | 43.66 ± 1.89 | 4.33 | 87.32 | 50.59 ± 6.72 | 13.28 | 101.18 | |
| 250 | 222.27 ± 2.77 | 1.25 | 88.91 | 270.32 ± 33.37 | 12.34 | 108.13 | |
| Diclofenac | 5 | 5.05 ± 0.42 | 8.35 | 100.90 | 5.17 ± 0.55 | 10.73 | 103.34 |
| 50 | 47.90 ± 0.38 | 0.79 | 95.80 | 47.89 ± 5.28 | 11.02 | 95.77 | |
| 250 | 248.34 ± 20.03 | 8.07 | 99.34 | 244.29 ± 16.02 | 6.56 | 97.72 | |
| 4-Hydroxy diclofenac | 5 | 4.54 ± 0.31 | 6.91 | 90.76 | 4.68 ± 0.43 | 9.24 | 93.51 |
| 50 | 49.19 ± 1.59 | 3.23 | 98.38 | 47.95 ± 5.19 | 10.83 | 95.90 | |
| 250 | 261.64 ± 18.42 | 7.04 | 104.66 | 271.16 ± 24.18 | 8.92 | 108.47 | |
| Omeprazole | 5 | 4.46 ± 0.07 | 1.65 | 89.27 | 5.33 ± 0.65 | 12.15 | 106.60 |
| 50 | 52.39 ± 1.04 | 1.99 | 104.79 | 53.48 ± 4.79 | 8.96 | 106.96 | |
| 250 | 270.27 ± 3.83 | 1.42 | 108.11 | 276.90 ± 18.04 | 6.52 | 110.76 | |
| 5-Hydroxy omeprazole | 5 | 4.53 ± 0.10 | 2.11 | 90.61 | 5.27 ± 0.59 | 11.12 | 105.32 |
| 50 | 51.48 ± 2.96 | 5.76 | 102.96 | 52.29 ± 4.44 | 8.49 | 104.57 | |
| 250 | 274.62 ± 2.60 | 0.95 | 109.85 | 269.70 ± 21.81 | 8.09 | 107.88 | |
| Nifedipine | 5 | 4.52 ± 0.31 | 6.89 | 90.38 | 4.43 ± 0.36 | 8.04 | 88.59 |
| 50 | 50.39 ± 0.66 | 1.31 | 100.79 | 46.45 ± 5.28 | 11.37 | 92.89 | |
| 250 | 262.14 ± 22.27 | 8.49 | 104.85 | 254.12 ± 21.95 | 8.64 | 101.65 | |
| Dehydronifedipine | 5 | 4.52 ± 0.40 | 8.94 | 90.32 | 4.62 ± 0.39 | 8.48 | 92.45 |
| 50 | 48.02 ± 2.79 | 5.81 | 96.04 | 50.67 ± 1.28 | 2.52 | 101.33 | |
| 250 | 239.74 ± 11.09 | 4.63 | 95.90 | 272.04 ± 18.83 | 6.92 | 108.81 | |
| Dextromethorphan | 5 | 4.50 ± 0.22 | 4.90 | 90.02 | 4.77 ± 0.33 | 6.91 | 95.46 |
| 50 | 50.88 ± 1.25 | 2.47 | 101.76 | 49.75 ± 4.22 | 8.47 | 99.50 | |
| 250 | 269.65 ± 11.78 | 4.37 | 107.86 | 266.69 ± 20.50 | 7.69 | 106.68 | |
| Dextrorphan | 5 | 4.43 ± 0.04 | 0.94 | 88.59 | 5.17 ± 0.54 | 10.36 | 103.43 |
| 50 | 52.82 ± 5.04 | 9.53 | 105.65 | 52.13 ± 5.20 | 9.97 | 104.27 | |
| 250 | 276.68 ± 6.37 | 2.30 | 110.67 | 278.23 ± 28.29 | 10.17 | 111.29 | |
| Metformin | 5 | 4.98 ± 0.19 | 3.82 | 99.59 | 5.25 ± 0.28 | 5.38 | 105.09 |
| 50 | 51.78 ± 2.29 | 4.43 | 103.55 | 49.67 ± 5.39 | 10.85 | 99.33 | |
| 250 | 256.67 ± 11.28 | 4.39 | 102.67 | 235.72 ± 7.66 | 3.25 | 94.29 | |
| Furosemide | 5 | 4.77 ± 0.46 | 9.55 | 95.43 | 4.59 ± 0.48 | 10.47 | 91.89 |
| 50 | 45.98 ± 2.29 | 4.99 | 91.97 | 44.47 ± 2.55 | 5.74 | 88.95 | |
| 250 | 247.19 ± 9.61 | 3.89 | 98.88 | 242.15 ± 8.51 | 3.51 | 96.86 | |
| Valsartan | 5 | 4.85 ± 0.45 | 9.23 | 97.03 | 4.87 ± 0.46 | 9.48 | 97.31 |
| 50 | 49.07 ± 0.88 | 1.80 | 98.14 | 46.68 ± 4.90 | 10.51 | 93.35 | |
| 250 | 270.50 ± 5.55 | 2.05 | 108.20 | 249.08 ± 20.98 | 8.42 | 99.63 | |
| Digoxin | 5 | 4.95 ± 0.06 | 1.20 | 99.00 | 4.98 ± 0.03 | 0.59 | 99.65 |
| 50 | 44.16 ± 2.58 | 5.83 | 88.32 | 45.15 ± 2.73 | 6.06 | 90.29 | |
| 250 | 245.12 ± 13.38 | 5.46 | 98.05 | 248.52 ± 22.38 | 9.01 | 99.41 | |
| Methotrexate | 5 | 5.00 ± 0.57 | 11.38 | 100.05 | 5.03 ± 0.57 | 11.39 | 100.59 |
| 50 | 52.90 ± 4.62 | 8.74 | 105.80 | 45.11 ± 5.41 | 11.99 | 90.22 | |
| 250 | 270.17 ± 9.54 | 3.53 | 108.07 | 251.27 ± 21.74 | 8.65 | 100.51 | |
| Analyte | Spiked Concentration (ng/mL) | Short Term Stability (4 h, 25 °C) | Free-Thaw Cycle Stability (−80 °C/25 °C, 3 Cycles) | Post Treatment Stability (6 °C, 24 h) |
|---|---|---|---|---|
| Caffeine | 5 | 96.9 ± 7.9 | 91.7 ± 10.7 | 92.4 ± 12.9 |
| 250 | 105.3 ± 8.2 | 108.5 ± 7.7 | 109.2 ± 12.4 | |
| Paraxanthine | 5 | 90.7 ± 9.8 | 80.6 ± 21.9 | 82.0 ± 15.7 |
| 250 | 95.6 ± 3.9 | 107.8 ± 7.4 | 112.0 ± 10.5 | |
| Diclofenac | 5 | 102.6 ± 10.5 | 108.3 ± 21.4 | 92.2 ± 14.6 |
| 250 | 106.6 ± 10.7 | 102.3 ± 2.6 | 103.2 ± 3.3 | |
| 4-Hydroxy diclofenac | 5 | 98.1 ± 6.5 | 107.2 ± 26.2 | 91.6 ± 12.9 |
| 250 | 100.4 ± 0.5 | 94.7 ± 5.6 | 108.8 ± 11.9 | |
| Omeprazole | 5 | 101.3 ± 5.4 | 100.2 ± 17.8 | 96.5 ± 18.3 |
| 250 | 105.0 ± 8.7 | 100.9 ± 4.9 | 109.4 ± 10.5 | |
| 5-Hydroxy omeprazole | 5 | 103.1 ± 3.1 | 101.8 ± 17.0 | 98.3 ± 7.4 |
| 250 | 99.6 ± 2.8 | 104.6 ± 4.0 | 111.2 ± 9.8 | |
| Nifedipine | 5 | 98.0 ± 1.8 | 94.4 ± 10.8 | 98.6 ± 7.3 |
| 250 | 99.7 ± 1.9 | 109.5 ± 19.4 | 106.5 ± 16.3 | |
| Dehydronifedipine | 5 | 96.2 ± 5.6 | 86.9 ± 12.5 | 91.6 ± 14.3 |
| 250 | 103.1 ± 6.4 | 107.5 ± 14.9 | 111.8 ± 10.9 | |
| Dextromethorphan | 5 | 94.8 ± 4.7 | 97.3 ± 9.0 | 91.1 ± 11.3 |
| 250 | 101.7 ± 2.4 | 101.5 ± 4.0 | 114.5 ± 12.9 | |
| Dextrorphan | 5 | 105.1 ± 5.7 | 98.9 ± 9.3 | 98.1 ± 10.2 |
| 250 | 110.3 ± 9.7 | 96.6 ± 3.7 | 99.5 ± 4.5 | |
| Metformin | 5 | 96.4 ± 18.2 | 104.8 ± 4.5 | 100.0 ± 3.7 |
| 250 | 102.7 ± 6.8 | 106.5 ± 15.5 | 102.9 ± 14.2 | |
| Furosemide | 5 | 98.4 ± 2.1 | 90.1 ± 9.2 | 89.7 ± 8.9 |
| 250 | 101.3 ± 1.2 | 107.4 ± 18.4 | 105.5 ± 14.2 | |
| Valsartan | 5 | 91.6 ± 14.3 | 101.5 ± 1.3 | 104.5 ± 5.1 |
| 250 | 102.8 ± 3.5 | 103.9 ± 20.1 | 101.2 ± 12.9 | |
| Digoxin | 5 | 97.9 ± 3.2 | 89.6 ± 9.2 | 99.1 ± 21.0 |
| 250 | 96.9 ± 3.8 | 102.6 ± 16.4 | 97.8 ± 7.0 | |
| Methotrexate | 5 | 96.6 ± 4.1 | 98.1 ± 19.4 | 92.6 ± 6.4 |
| 250 | 102.7 ± 4.6 | 106.4 ± 21.3 | 109.7 ± 18.1 |
| Cyps | Probe Substrate | Parameters | Single (n = 6) | CYP Cocktail (n = 6) |
|---|---|---|---|---|
| Cyp1a2 | Caffeine | Cmax (ng/mL) | 866.9 ± 310.8 | 739.5 ± 216.8 |
| AUC24 h (ng·h/mL) | 3042.8 ± 1226.5 | 3965.1 ± 1632.2 | ||
| AUC∞ (ng·h/mL) | 3051.3 ± 1231.0 | 3991.4 ± 1661.3 | ||
| Paraxanthine | AUC24 h (ng·h/mL) | 2878.6 ± 931.3 | 3593.8 ± 1119.0 | |
| AUC∞ (ng·h/mL) | 3135.4 ± 985.6 | 3152.4 ± 471.4 | ||
| MR | 1.1 ± 0.4 | 1.0 ± 0.4 | ||
| Cyp2c11 | Diclofenac | Cmax (ng/mL) | 1863.9 ± 1309.8 | 1559.1 ± 1036.2 |
| AUC24 h (ng·h/mL) | 4386.5 ± 1264.3 | 5505.2 ± 1807.6 | ||
| AUC∞ (ng·h/mL) | 4552.2 ± 1349.0 | 5741.9 ± 1836.8 | ||
| 4-Hydroxy diclofenac | AUC24 h (ng·h/mL) | 4080.0 ± 2217.4 | 6944.7 ± 3761.8 | |
| AUC∞ (ng·h/mL) | 5831.5 ± 3529.0 | 7844.7 ± 4354.1 | ||
| MR | 1.0 ± 0.6 | 1.2 ± 0.3 | ||
| Cyp2c11 | Omeprazole | Cmax (ng/mL) | 182.3 ± 158.5 | 62.0 ± 28.0 |
| AUC24 h (ng·h/mL) | 134.3 ± 107.6 | 145.1 ± 87.0 | ||
| AUC∞ (ng·h/mL) | 134.5 ± 107.8 | 145.6 ± 87.0 | ||
| 5-Hydroxy omeprazole | AUC24 h (ng·h/mL) | 116.6 ± 69.7 | 110.9 ± 63.8 | |
| AUC∞ (ng·h/mL) | 119.0 ± 71.4 | 112.0 ± 64.6 | ||
| MR | 1.0 ± 0.2 | 0.8 ± 0.1 | ||
| Cyp3a1 | Nifedipine | Cmax (ng/mL) | 445.8 ± 202.0 | 420.1 ± 224.5 |
| AUC24 h (ng·h/mL) | 1392.2 ± 397.5 | 1490.9 ± 844.5 | ||
| AUC∞ (ng·h/mL) | 1393.8 ± 397.0 | 1499.6 ± 841.9 | ||
| Dehydronifedipine | AUC24 h (ng·h/mL) | 22.3 ± 5.8 | 37.0 ± 15.8 | |
| AUC∞ (ng·h/mL) | 22.6 ± 5.8 | 37.4 ± 15.9 | ||
| MR | 0.018 ± 0.01 | 0.028 ± 0.01 | ||
| Cyp2d2 | Dextromethorphan | Cmax (ng/mL) | 117.9 ± 78.1 | 180.0 ± 136.2 |
| AUC24 h (ng·h/mL) | 457.5 ± 373.3 | 971.1 ± 626.2 | ||
| AUC∞ (ng·h/mL) | 459.1 ± 374.4 | 1011.1 ± 625.8 | ||
| Dextrorphan | AUC24 h (ng·h/mL) | 142.3 ± 137.0 | 196.9 ± 214.6 | |
| AUC∞ (ng·h/mL) | 208.6 ± 145.3 | 196.0 ± 121.9 | ||
| MR | 0.30 ± 0.16 | 0.22 ± 0.20 |
| Transporters | Probe Substrate | Parameters | Single (n = 6) | Transporter Cocktail (n = 6) |
|---|---|---|---|---|
| Oct1/2 | Metformin | AUC24 h (ng·h/mL) | 733 ± 402 | 767 ± 245 |
| AUC∞ (ng·h/mL) | 750 ± 415 | 777 ± 247 | ||
| Xu (ng) | 47,806 ± 29,071 | 52,474 ± 19,441 | ||
| Oat1/3 | Furosemide | AUC24 h (ng·h/mL) | 766 ± 370 | 652 ± 130 |
| AUC∞ (ng·h/mL) | 773 ± 366 | 654 ± 130 | ||
| Xu (ng) | 2330 ± 1285 | 6969 ± 3330 | ||
| Oatp2 | Valsartan | AUC24 h (ng·h/mL) | 2252 ± 247 | 2198 ± 518 |
| AUC∞ (ng·h/mL) | 2285 ± 245 | 2225 ± 519 | ||
| Xu (ng) | 920 ± 504 | 627 ± 441 | ||
| P-gp | Digoxin | AUC24 h (ng·h/mL) | 4079 ± 716 | 3151 ± 1009 |
| AUC∞ (ng·h/mL) | 4232 ± 828 | 3193 ± 1011 | ||
| Xu (ng) | 29,954 ± 11,498 | 23,444 ± 10,659 | ||
| Mrp2 | Methotrexate | AUC24 h (ng·h/mL) | 1339 ± 242 | 1319 ± 252 |
| AUC∞ (ng·h/mL) | 1362 ± 264 | 1329 ± 260 | ||
| Xu (ng) | 2604 ± 1924 | 4621 ± 1431 |
| Cyps/Transporters | Probe Substrate | Parameters | CYP or Transporter Cocktail (n = 6) | Dual Cocktail (n = 7) |
|---|---|---|---|---|
| Cyp1a2 | Caffeine | AUC24 h (ng·h/mL) | 3965 ± 1632 | 3533 ± 1788 |
| AUC∞ (ng·h/mL) | 3991 ± 1661 | 3580 ± 1889 | ||
| Paraxanthine | AUC24 h (ng·h/mL) | 3593.83 ± 1119.05 | 2416.56 ± 1073.93 | |
| AUC∞ (ng·h/mL) | 3152.35 ± 471.42 | 2573.44 ± 1125.72 | ||
| MR | 1.01 ± 0.37 | 0.77 ± 0.17 | ||
| Cyp2c11 | Diclofenac | AUC24 h (ng·h/mL) | 5505 ± 1808 | 5073 ± 1186 |
| AUC∞ (ng·h/mL) | 5742 ± 1837 | 5116 ± 1200 | ||
| 4-Hydroxy diclofenac | AUC24 h (ng·h/mL) | 6944.71 ± 3761.81 | 5184.02 ± 1609.43 | |
| AUC∞ (ng·h/mL) | 7844.69 ± 4354.05 | 5707.53 ± 2017.07 | ||
| MR | 1.20 ± 0.34 | 1.02 ± 0.13 | ||
| Cyp2c11 | Omeprazole | AUC24 h (ng·h/mL) | 145 ± 87 | 191 ± 53 |
| AUC∞ (ng·h/mL) | 146 ± 87 | 194 ± 52 | ||
| 5-Hydroxyomeprazole | AUC24 h (ng·h/mL) | 110.93 ± 63.79 | 147.55 ± 68.57 | |
| AUC∞ (ng·h/mL) | 111.99 ± 64.62 | 150.42 ± 67.29 | ||
| MR | 0.76 ± 0.08 | 0.74 ± 0.14 | ||
| Cyp3a4 | Nifedipine | AUC24 h (ng·h/mL) | 1491 ± 844 | 1320 ± 206 |
| AUC∞ (ng·h/mL) | 1500 ± 842 | 1343 ± 204 | ||
| Dehydronifedipine | AUC24 h (ng·h/mL) | 37.03 ± 15.84 | 35.02 ± 10.94 | |
| AUC∞ (ng·h/mL) | 37.37 ± 15.90 | 36.99 ± 10.69 | ||
| MR | 0.028 ± 0.012 | 0.025 ± 0.008 | ||
| Cyp2d2 | Dextromethorphan | AUC24 h (ng·h/mL) | 971 ± 626 | 654 ± 406 |
| AUC∞ (ng·h/mL) | 1011 ± 626 | 674 ± 387 | ||
| Dextrorphan | AUC24 h (ng·h/mL) | 196.90 ± 214.62 | 155.63 ± 69.83 | |
| AUC∞ (ng·h/mL) | 196.02 ± 121.90 | 213.90 ± 74.20 | ||
| MR | 0.20 ± 0.22 | 0.26 ± 0.08 | ||
| Oct1/2 | Metformin | AUC24 h (ng·h/mL) | 767 ± 245 | 1027 ± 349 |
| AUC∞ (ng·h/mL) | 777 ± 247 | 1032 ± 348 | ||
| Xu (ng) | 52,474 ± 19,441 | 48,885 ± 17,369 | ||
| Oat1/3 | Furosemide | AUC24 h (ng·h/mL) | 652 ± 130 | 701 ± 175 |
| AUC∞ (ng·h/mL) | 654 ± 130 | 704 ± 174 | ||
| Xu (ng) | 6969 ± 3330 | 4950 ± 2211 | ||
| Oatp2 | Valsartan | AUC24 h (ng·h/mL) | 2198 ± 518 | 2258 ± 674 |
| AUC∞ (ng·h/mL) | 2225 ± 519 | 2289 ± 667 | ||
| Xu (ng) | 627 ± 441 | 516 ± 338 | ||
| P-gp | Digoxin | AUC24 h (ng·h/mL) | 3151 ± 1009 | 3302 ± 717 |
| AUC∞ (ng·h/mL) | 3193 ± 1011 | 3430 ± 727 | ||
| Xu (ng) | 23,444 ± 10,659 | 18,542 ± 7040 | ||
| Mrp2 | Methotrexate | AUC24 h (ng·h/mL) | 1319 ± 252 | 1432 ± 414 |
| AUC∞ (ng·h/mL) | 1329 ± 260 | 1451 ± 421 | ||
| Xu (ng) | 4621 ± 1431 | 5680 ± 3451 |
| Cyps/Transporters | Probe Substrate | Parameters | Inhibition | Induction | |||
|---|---|---|---|---|---|---|---|
| Control (n = 6) | Ketoconazole (n = 4) | Rifampin (n = 4) | Control (n = 4) | Rifampin (n = 4) | |||
| Cyp1a2 | Caffeine | AUC24 h (ng·h/mL) | 3104.8 ± 963.8 | 3579.8 ± 891.5 | 4131.9 ± 2129.2 | 3959.0 ± 1824.8 | 3687.9 ± 540.0 |
| Paraxanthine | AUC24 h (ng·h/mL) | 2416.6 ± 1073.9 | 3138.1 ± 1042.8 | 3555.2 ± 1740.2 | 3248.6 ± 819.6 | 3107.9 ± 351.6 | |
| MR | 0.8 ± 0.2 | 0.9 ± 0.1 | 0.9 ± 0.0 | 0.9 ± 0.4 | 0.9 ± 0.1 | ||
| Cyp2c11 | Diclofenac | AUC24 h (ng·h/mL) | 3551.7 ± 887.7 | 3863.3 ± 1109.8 | 3681.9 ± 724.2 | 4980.0 ± 1419.7 | 5744.9 ± 1647.1 |
| 4-Hydroxy diclofenac | AUC24 h (ng·h/mL) | 3480.8 ± 1062.3 | 3584.7 ± 1222.2 | 3024.5 ± 459.7 | 5848.2 ± 2944.7 | 3986.8 ± 5627.2 | |
| MR | 1.0 ± 0.2 | 0.9 ± 0.2 | 0.8 ± 0.2 | 1.1 ± 0.3 | 0.6 ± 0.6 | ||
| Cyp2c11 | Omeprazole | AUC24 h (ng·h/mL) | 191.3 ± 53.3 | 403.5 ± 116.0 * | 217.4 ± 18.3 | 207.9 ± 43.1 | 245.2 ± 31.2 |
| 5-Hydroxyomeprazole | AUC24 h (ng·h/mL) | 119.0 ± 27.5 | 121.4 ± 37.1 | 105.0 ± 24.6 | 129.0 ± 17.8 | 133.4 ± 26.9 | |
| MR | 0.6 ± 0.1 | 0.3 ± 0.0 * | 0.5 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 | ||
| Cyp3a1 | Nifedipine | AUC24 h (ng·h/mL) | 1367.3 ± 201.1 | 5021.9 ± 3577.3 * | 1597.3 ± 187.6 | 1595.2 ± 648.7 | 859.0 ± 268.2 * |
| Dehydronifedipine | AUC24 h (ng·h/mL) | 60.2 ± 16.4 | 39.2 ± 11.1 * | 67.5 ± 4.6 | 40.6 ± 14.7 | 56.0 ± 13.1 | |
| MR | 0.05 ± 0.02 | 0.01 ± 0.00 * | 0.04 ± 0.00 | 0.0 ± 0.0 | 0.1 ± 0.0 * | ||
| Cyp2d2 | Dextromethorphan | AUC24 h (ng·h/mL) | 1002.4 ± 206.0 | 1170.3 ± 228.8 | 1029.7 ± 455.1 | 1117.4 ± 574.1 | 970.1 ± 188.4 |
| Dextrorphan | AUC24 h (ng·h/mL) | 234.5 ± 167.5 | 142.5 ± 27.0 | 230.6 ± 43.1 | 229.2 ± 223.0 | 361.9 ± 205.8 | |
| MR | 0.2 ± 0.2 | 0.1 ± 0.0 | 0.2 ± 0.1 | 0.2 ± 0.2 | 0.4 ± 0.2 | ||
| Oct1/2 | Metformin | AUC24 h (ng·h/mL) | 908.0 ± 159.7 | 980.2 ± 90.3 | 788.1 ± 127.4 | 976.3 ± 406.4 | 1294.9 ± 468.0 |
| Oat1/3 | Furosemide | AUC24 h (ng·h/mL) | 700.9 ± 175.2 | 731.5 ± 123.6 | 682.6 ± 76.2 | 614.5 ± 83.9 | 524.2 ± 28.2 |
| Oatp2 | Valsartan | AUC24 h (ng·h/mL) | 2488.5 ± 315.6 | 2227.7 ± 1007.3 | 8693.9 ± 936.1 * | 2258.4 ± 673.5 | 2333.4 ± 137.5 |
| P-gp | Digoxin | AUC24 h (ng·h/mL) | 3302.0 ± 717.5 | 3677.2 ± 745.8 | 3732.4 ± 220.8 | 3302.0 ± 717.5 | 1349.5 ± 738.9 * |
| Mrp2 | Methotrexate | AUC24 h (ng·h/mL) | 1432.1 ± 413.8 | 1229.0 ± 498.4 | 1332.9 ± 463.9 | 1432.1 ± 413.8 | 1294.0 ± 484.0 |
| Category | Isoforms in Rat (Human) | Probe Substrates (Dose, mg/kg) | Route of Administration | Phenotypic Index |
|---|---|---|---|---|
| CYPs | Cyp1a2 (CYP1A2) | Caffeine (1 mg/kg) | PO | AUC |
| Cyp2c11 (CYP2C9) | Diclofenac (2 mg/kg) | AUC | ||
| Cyp2c11 (CYP2C19) | Omeprazole (2 mg/kg) | AUC | ||
| Cyp2d2 (CYP2D6) | Dextromethorphan (10 mg/kg) | AUC | ||
| Cyp3a1 (CYP3A4) | Nifedipine (0.5 mg/kg) | AUC | ||
| Transporters | Oct1/2 (OCT1/2) | Metformin (0.5 mg/kg) | IV | AUC |
| Oat1/3 (OAT1/3) | Furosemide (0.1 mg/kg) | AUC | ||
| Oatp2 (OATP1B1/1B3) | Valsartan (0.2 mg/kg) | AUC | ||
| P-gp (P-gp) | Digoxin (2 mg/kg) | AUC | ||
| Mrp2 (MRP2) | Methotrexate (0.5 mg/kg) | AUC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, M.; Jeon, J.-H.; Choi, M.-K.; Song, I.-S. The Development and Validation of a Novel “Dual Cocktail” Probe for Cytochrome P450s and Transporter Functions to Evaluate Pharmacokinetic Drug-Drug and Herb-Drug Interactions. Pharmaceutics 2020, 12, 938. https://doi.org/10.3390/pharmaceutics12100938
Kwon M, Jeon J-H, Choi M-K, Song I-S. The Development and Validation of a Novel “Dual Cocktail” Probe for Cytochrome P450s and Transporter Functions to Evaluate Pharmacokinetic Drug-Drug and Herb-Drug Interactions. Pharmaceutics. 2020; 12(10):938. https://doi.org/10.3390/pharmaceutics12100938
Chicago/Turabian StyleKwon, Mihwa, Ji-Hyeon Jeon, Min-Koo Choi, and Im-Sook Song. 2020. "The Development and Validation of a Novel “Dual Cocktail” Probe for Cytochrome P450s and Transporter Functions to Evaluate Pharmacokinetic Drug-Drug and Herb-Drug Interactions" Pharmaceutics 12, no. 10: 938. https://doi.org/10.3390/pharmaceutics12100938