Effectiveness of a Pharmacogenetic Tool at Improving Treatment Efficacy in Major Depressive Disorder: A Meta-Analysis of Three Clinical Studies

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collected
- Design and duration of each study;
- Patients’ characteristics as per treatment group: sample size, mean age, gender distribution, ethnicity, psychiatric comorbidities, baseline and final visit CGI-S scores, and baseline and final visit HDRS-17 scores when available.
2.2. Outcomes
2.3. Statistics
2.4. Assessment of Bias
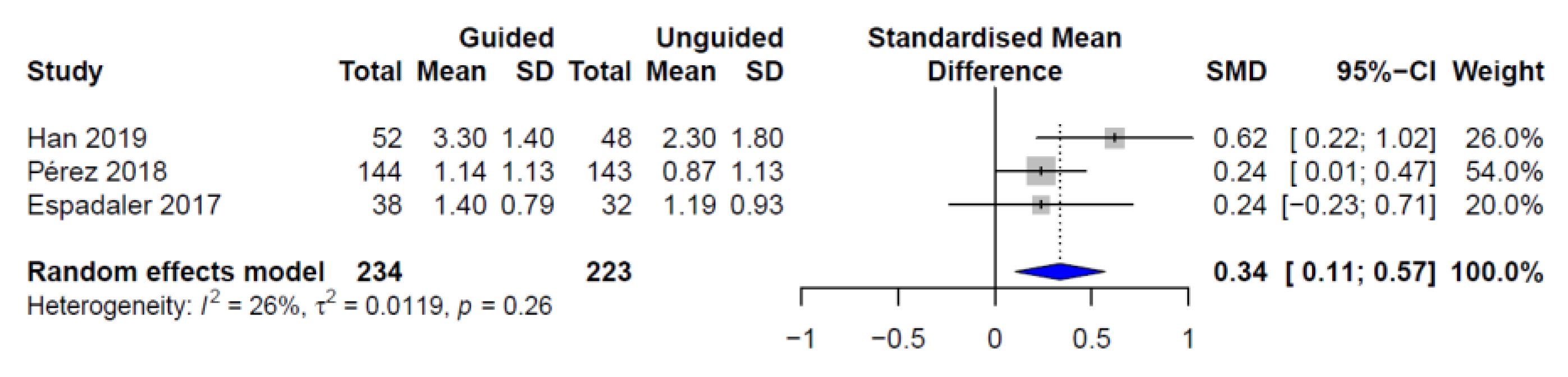
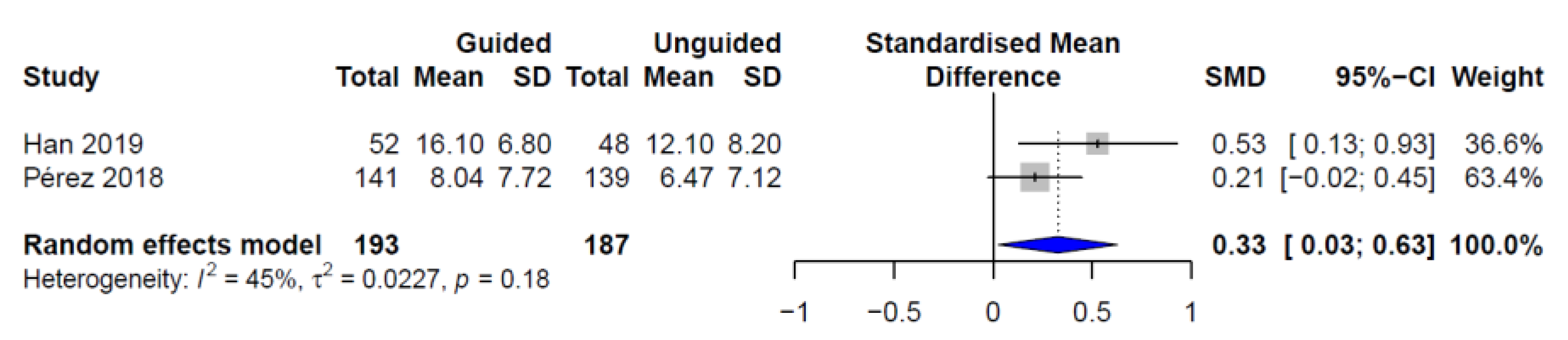
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Depression and Other Common Mental Disorders; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Steel, Z.; Marnane, C.; Iranpour, C.; Chey, T.; Jackson, J.W.; Patel, V.; Silove, D. The global prevalence of common mental disorders: A systematic review and meta-analysis 1980–2013. Int. J. Epidemiol. 2014, 43, 476–493. [Google Scholar] [CrossRef]
- König, H.; König, H.-H.; Konnopka, A. The excess costs of depression: A systematic review and meta-analysis. Epidemiol. Psychiatr. Sci. 2019, 1–16. [Google Scholar] [CrossRef]
- Vos, T.; Abajobir, A.A.; Abate, K.H.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abdulkader, R.S.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef]
- Rush, A.J.; Trivedi, M.H.; Wisniewski, S.R.; Nierenberg, A.A.; Stewart, J.W.; Warden, D.; Niederehe, G.; Thase, M.E.; Lavori, P.W.; Lebowitz, B.D.; et al. Acute and Longer-Term Outcomes in Depressed Outpatients Requiring One or Several Treatment Steps: A STAR*D Report. Am. J. Psychiatry 2006, 163, 1905–1917. [Google Scholar] [CrossRef]
- Fournier, J.C.; DeRubeis, R.J.; Hollon, S.D.; Dimidjian, S.; Amsterdam, J.D.; Shelton, R.C.; Fawcett, J. Antidepressant drug effects and depression severity: A patient-level meta-analysis. JAMA 2010, 303, 47–53. [Google Scholar] [CrossRef]
- Johnston, K.M.; Powell, L.C.; Anderson, I.M.; Szabo, S.; Cline, S. The burden of treatment-resistant depression: A systematic review of the economic and quality of life literature. J. Affect. Disord. 2019, 242, 195–210. [Google Scholar] [CrossRef]
- Sheehan, D.V.; Keene, M.S.; Eaddy, M.; Krulewicz, S.; Kraus, J.E.; Carpenter, D.J. Differences in medication adherence and healthcare resource utilization patterns: Older versus newer antidepressant agents in patients with depression and/or anxiety disorders. CNS Drugs 2008, 22, 963–973. [Google Scholar] [CrossRef]
- Warden, D.; Rush, A.J.; Trivedi, M.H.; Fava, M.; Wisniewski, S.R. The STAR*D Project results: A comprehensive review of findings. Curr. Psychiatry Rep. 2007, 9, 449–459. [Google Scholar] [CrossRef]
- Kelly, K.; Posternak, M.; Alpert, J.E. Toward achieving optimal response: Understanding and managing antidepressant side effects. Dialogues Clin. Neurosci. 2008, 10, 409–418. [Google Scholar]
- Swen, J.J.; Nijenhuis, M.; de Boer, A.; Grandia, L.; Maitland-van der Zee, A.H.; Mulder, H.; Rongen, G.A.; van Schaik, R.H.; Schalekamp, T.; Touw, D.J.; et al. Pharmacogenetics: From bench to byte—An update of guidelines. Clin. Pharmacol. Ther. 2011, 89, 662–673. [Google Scholar] [CrossRef]
- Leckband, S.G.; Kelsoe, J.R.; Dunnenberger, H.M.; George, A.L., Jr.; Tran, E.; Berger, R.; Muller, D.J.; Whirl-Carrillo, M.; Caudle, K.E.; Pirmohamed, M. Clinical Pharmacogenetics Implementation Consortium Guidelines for HLA-B Genotype and Carbamazepine Dosing. Clin. Pharmacol. Ther. 2013, 94, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Caudle, K.E.; Rettie, A.E.; Whirl-Carrillo, M.; Smith, L.H.; Mintzer, S.; Lee, M.T.; Klein, T.E.; Callaghan, J.T. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and HLA-B Genotypes and Phenytoin Dosing. Clin. Pharmacol. Ther. 2014, 96, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Hicks, J.K.; Sangkuhl, K.; Swen, J.J.; Ellingrod, V.L.; Muller, D.J.; Shimoda, K.; Bishop, J.R.; Kharasch, E.D.; Skaar, T.C.; Gaedigk, A.; et al. Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin. Pharmacol. Ther. 2017, 102, 37–44. [Google Scholar] [CrossRef]
- Hicks, J.K.; Bishop, J.R.; Sangkuhl, K.; Muller, D.J.; Ji, Y.; Leckband, S.G.; Leeder, J.S.; Graham, R.L.; Chiulli, D.L.; LLerena, A.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin. Pharmacol. Ther. 2015, 98, 127–134. [Google Scholar] [CrossRef]
- Caudle, K.E.; Keeling, N.J.; Klein, T.E.; Whirl-Carrillo, M.; Pratt, V.M.; Hoffman, J.M. Standardization can accelerate the adoption of pharmacogenomics: Current status and the path forward. Pharmacogenomics 2018, 19, 847–860. [Google Scholar] [CrossRef]
- Bank, P.C.D.; Caudle, K.E.; Swen, J.J.; Gammal, R.S.; Whirl-Carrillo, M.; Klein, T.E.; Relling, M.V.; Guchelaar, H.J. Comparison of the Guidelines of the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetics Working Group. Clin. Pharmacol. Ther. 2018. [Google Scholar] [CrossRef]
- Drozda, K.; Muller, D.J.; Bishop, J.R. Pharmacogenomic testing for neuropsychiatric drugs: Current status of drug labeling, guidelines for using genetic information, and test options. Pharmacotherapy 2014, 34, 166–184. [Google Scholar] [CrossRef]
- Ehmann, F.; Caneva, L.; Prasad, K.; Paulmichl, M.; Maliepaard, M.; Llerena, A.; Ingelman-Sundberg, M.; Papaluca-Amati, M. Pharmacogenomic information in drug labels: European Medicines Agency perspective. Pharmacogenom. J. 2015, 15, 201. [Google Scholar] [CrossRef]
- Haddow, J.E.; Palomaki, G.E. ACCE: A Model Process for Evaluating Data on Emerging Genetic Tests. In Human Genome Epidemiology: A Scientific Foundation for Using Genetic Information to Improve Health and Prevent Disease; Khoury, M.J., Little, J., Burke, W., Eds.; Oxford University Press: Oxford, UK, 2004; pp. 217–233. ISBN 0195146743. [Google Scholar]
- Bousman, C.A.; Hopwood, M. Commercial pharmacogenetic-based decision-support tools in psychiatry. Lancet Psychiatry 2016, 3, 585–590. [Google Scholar] [CrossRef]
- Wisniewski, S.R.; Rush, A.J.; Nierenberg, A.A.; Gaynes, B.N.; Warden, D.; Luther, J.F.; McGrath, P.J.; Lavori, P.W.; Thase, M.E.; Fava, M.; et al. Can phase III trial results of antidepressant medications be generalized to clinical practice? A STAR*D report. Am. J. Psychiatry 2009, 166, 599–607. [Google Scholar] [CrossRef]
- Perez, V.; Salavert, A.; Espadaler, J.; Tuson, M.; Saiz-Ruiz, J.; Saez-Navarro, C.; Bobes, J.; Baca-Garcia, E.; Vieta, E.; Olivares, J.M.; et al. Efficacy of prospective pharmacogenetic testing in the treatment of major depressive disorder: Results of a randomized, double-blind clinical trial. BMC Psychiatry 2017, 17, 250. [Google Scholar] [CrossRef]
- Han, C.; Wang, S.-M.; Bahk, W.-M.; Lee, S.-J.; Patkar, A.A.; Masand, P.S.; Mandelli, L.; Pae, C.-U.; Serretti, A. A Pharmacogenomic-based Antidepressant Treatment for Patients with Major Depressive Disorder: Results from an 8-week, Randomized, Single-blinded Clinical Trial. Clin. Psychopharmacol. Neurosci. 2018, 16, 469–480. [Google Scholar] [CrossRef]
- Espadaler, J.; Tuson, M.; Lopez-Ibor, J.M.; Lopez-Ibor, F.; Lopez-Ibor, M.I. Pharmacogenetic testing for the guidance of psychiatric treatment: A multicenter retrospective analysis. CNS Spectr. 2017, 22, 315–324. [Google Scholar] [CrossRef]
- Guy, W. ECDEU Assessment Manual for Psychopharmacology: Revised; DHEW Publication No. ADM 76–338; US Department of Health, Education and Welfare: Rockville, MD, USA, 1976. [Google Scholar]
- Hamilton, M. A rating scale for depression. J. Neurol. Neurosurg. Psychiatry 1960, 23, 56. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gotzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: Explanation and elaboration. BMJ 2009, 339, b2700. [Google Scholar] [CrossRef]
- Porcelli, S.; Fabbri, C.; Serretti, A. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with antidepressant efficacy. Eur. Neuropsychopharmacol. 2012, 22, 239–258. [Google Scholar] [CrossRef]
- Niitsu, T.; Fabbri, C.; Bentini, F.; Serretti, A. Pharmacogenetics in major depression: A comprehensive meta-analysis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 45C, 183–194. [Google Scholar] [CrossRef]
- Breitenstein, B.; Scheuer, S.; Bruckl, T.M.; Meyer, J.; Ising, M.; Uhr, M.; Holsboer, F. Association of ABCB1 gene variants, plasma antidepressant concentration, and treatment response: Results from a randomized clinical study. J. Psychiatr. Res. 2016, 73, 86–95. [Google Scholar] [CrossRef]
- Uhr, M.; Tontsch, A.; Namendorf, C.; Ripke, S.; Lucae, S.; Ising, M.; Dose, T.; Ebinger, M.; Rosenhagen, M.; Kohli, M.; et al. Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron 2008, 57, 203–209. [Google Scholar] [CrossRef]
- van der Weide, K.; van der Weide, J. The influence of the CYP3A4*22 polymorphism on serum concentration of quetiapine in psychiatric patients. J. Clin. Psychopharmacol. 2014, 34, 256–260. [Google Scholar] [CrossRef]
- Mas, S.; Gasso, P.; Ritter, M.A.; Malagelada, C.; Bernardo, M.; Lafuente, A. Pharmacogenetic predictor of extrapyramidal symptoms induced by antipsychotics: Multilocus interaction in the mTOR pathway. Eur. Neuropsychopharmacol. 2015, 25, 51–59. [Google Scholar] [CrossRef]
- Hedges, L. V Distribution Theory for Glass’s Estimator of Effect size and Related Estimators. J. Educ. Behav. Stat. 1981, 6, 107–128. [Google Scholar] [CrossRef]
- Schwarzer, G.; Carpenter, J.R.; Rücker, G. Meta-Analysis with R; Springer: New York, NY, USA, 2015; ISBN 9783319214153. [Google Scholar]
- Pishva, E.; Drukker, M.; Viechtbauer, W.; Decoster, J.; Collip, D.; van Winkel, R.; Wichers, M.; Jacobs, N.; Thiery, E.; Derom, C.; et al. Epigenetic genes and emotional reactivity to daily life events: A multi-step gene-environment interaction study. PLoS ONE 2014, 9, e100935. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2015. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Higgins, J.P.T.; Thompson, S.G.; Deeks, J.J.; Altman, D.G. Measuring inconsistency in meta-analyses. BMJ 2003, 327, 557–560. [Google Scholar] [CrossRef] [Green Version]
- Hasselblad, V.; Hedges, L.V. Meta-Analysis of Screening and Diagnostic Tests. Psychol. Bull. 1995. [Google Scholar] [CrossRef]
- Chinn, S. A simple method for converting an odds ratio to effect size for use in meta-analysis. Stat. Med. 2000, 19, 3127–3131. [Google Scholar] [CrossRef]
- Higgins, J.P.T.; Altman, D.G.; Gøtzsche, P.C.; Jüni, P.; Moher, D.; Oxman, A.D.; Savovic, J.; Schulz, K.F.; Weeks, L.; Sterne, J.A.C.; et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ 2011, 343, d5928. [Google Scholar] [CrossRef]
- Sterne, J.A.; Hernán, M.A.; Reeves, B.C.; Savović, J.; Berkman, N.D.; Viswanathan, M.; Henry, D.; Altman, D.G.; Ansari, M.T.; Boutron, I.; et al. ROBINS-I: A tool for assessing risk of bias in non-randomised studies of interventions. BMJ 2016, 355, i4919. [Google Scholar] [CrossRef]
- Gibertini, M.; Nations, K.R.; Whitaker, J.A. Obtained effect size as a function of sample size in approved antidepressants. Int. Clin. Psychopharmacol. 2012, 27, 100–106. [Google Scholar] [CrossRef]
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Chaimani, A.; Atkinson, L.Z.; Ogawa, Y.; Leucht, S.; Ruhe, H.G.; Turner, E.H.; Higgins, J.P.T.; et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: A systematic review and network meta-analysis. Lancet 2018, 391, 1357–1366. [Google Scholar] [CrossRef]
- Zimmerman, M.; Mattia, J.I.; Posternak, M.A. Are Subjects in Pharmacological Treatment Trials of Depression Representative of Patients in Routine Clinical Practice? Am. J. Psychiatry 2002, 159, 469–473. [Google Scholar] [CrossRef]
- Zetin, M.; Hoepner, C.T. Relevance of Exclusion Criteria in Antidepressant Clinical Trials. J. Clin. Psychopharmacol. 2007, 27, 295–301. [Google Scholar] [CrossRef]
- Blanco, C.; Olfson, M.; Goodwin, R.D.; Ogburn, E.; Liebowitz, M.R.; Nunes, E.V.; Hasin, D.S. Generalizability of clinical trial results for major depression to community samples: Results from the National Epidemiologic Survey on Alcohol and Related Conditions. J. Clin. Psychiatry 2008. [Google Scholar] [CrossRef]
- Kessler, R.C.; Berglund, P.; Demler, O.; Jin, R.; Koretz, D.; Merikangas, K.R.; Rush, A.J.; Walters, E.E.; Wang, P.S. National Comorbidity Survey Replication the Epidemiology of Major Depressive Disorder. JAMA 2003, 289, 3095–3105. [Google Scholar] [CrossRef]
- Hasin, D.S.; Goodwin, R.D.; Stinson, F.S.; Grant, B.F. Epidemiology of Major Depressive Disorder. Arch. Gen. Psychiatry 2005, 62, 1097–1106. [Google Scholar] [CrossRef]
- Zimmerman, M.; Chelminski, I.; McDermut, W. Major depressive disorder and axis I diagnostic comorbidity. J. Clin. Psychiatry 2002, 63, 187–193. [Google Scholar] [CrossRef]
- Melartin, T.K.; Rytsälä, H.J.; Leskelä, U.S.; Lestelä-Mielonen, P.S.; Sokero, T.P.; Isometsä, E.T. Current comorbidity of psychiatric disorders among DSM-IV major depressive disorder patients in psychiatric care in the Vantaa Depression Study. J. Clin. Psychiatry 2002, 63, 126–134. [Google Scholar] [CrossRef]
- Rush, A.J. STAR*D: What Have We Learned? Am. J. Psychiatry 2007, 164, 201–204. [Google Scholar] [CrossRef]
- Romera, I.; Pérez, V.; Menchón, J.M.; Polavieja, P.; Gilaberte, I. Optimal cutoff point of the Hamilton Rating Scale for Depression according to normal levels of social and occupational functioning. Psychiatry Res. 2011, 186, 133–137. [Google Scholar] [CrossRef]
- Cameron, I.M.; Reid, I.C.; MacGillivray, S.A. Efficacy and tolerability of antidepressants for sub-threshold depression and for mild major depressive disorder. J. Affect. Disord. 2014, 166, 48–58. [Google Scholar] [CrossRef]
- Singh, A.B. Improved Antidepressant Remission in Major Depression via a Pharmacokinetic Pathway Polygene Pharmacogenetic Report. Clin. Psychopharmacol. Neurosci. 2015, 13, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Bradley, P.; Shiekh, M.; Mehra, V.; Vrbicky, K.; Layle, S.; Olson, M.C.; Maciel, A.; Cullors, A.; Garces, J.A.; Lukowiak, A.A. Improved efficacy with targeted pharmacogenetic-guided treatment of patients with depression and anxiety: A randomized clinical trial demonstrating clinical utility. J. Psychiatr. Res. 2018, 96, 100–107. [Google Scholar] [CrossRef]
- Rush, A.J.; Fava, M.; Wisniewski, S.R.; Lavori, P.W.; Trivedi, M.H.; Sackeim, H.A.; Thase, M.E.; Nierenberg, A.A.; Quitkin, F.M.; Kashner, T.M.; et al. Sequenced treatment alternatives to relieve depression (STAR*D): Rationale and design. Control. Clin. Trials 2004, 25, 119–142. [Google Scholar] [CrossRef]
- Menchon, J.M.; Espadaler, J.; Tuson, M.; Saiz-Ruiz, J.; Bobes, J.; Vieta, E.; Alvarez, E.; Perez, V. Patient characteristics driving clinical utility in psychiatric pharmacogenetics: A reanalysis from the AB-GEN multicentric trial. J. Neural Transm. 2019. [Google Scholar] [CrossRef]
- Bousman, C.A.; Arandjelovic, K.; Mancuso, S.G.; Eyre, H.A.; Dunlop, B.W. Pharmacogenetic tests and depressive symptom remission: A meta-analysis of randomized controlled trials. Pharmacogenomics 2019, 20, 37–47. [Google Scholar] [CrossRef]
- Rosenblat, J.D.; Lee, Y.; McIntyre, R.S. The effect of pharmacogenomic testing on response and remission rates in the acute treatment of major depressive disorder: A meta-analysis. J. Affect. Disord. 2018, 241, 484–491. [Google Scholar] [CrossRef]
- Bousman, C.A.; Zierhut, H.; Müller, D.J. Navigating the Labyrinth of Pharmacogenetic Testing: A Guide to Test Selection. Clin. Pharmacol. Ther. 2019. [Google Scholar] [CrossRef]
- Bousman, C.A.; Dunlop, B.W. Genotype, phenotype, and medication recommendation agreement among commercial pharmacogenetic-based decision support tools. Pharmacogenom. J. 2018, 18, 613–622. [Google Scholar] [CrossRef]
- Zeier, Z.; Carpenter, L.L.; Kalin, N.H.; Rodriguez, C.I.; McDonald, W.M.; Widge, A.S.; Nemeroff, C.B. Clinical Implementation of Pharmacogenetic Decision Support Tools for Antidepressant Drug Prescribing. Am. J. Psychiatry 2018, 175, 873–886. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study | Study Design | Country | Sample Size | Demographics | Patient Characteristics |
|---|---|---|---|---|---|
| Korean study [24] |
| South Korea | 100 (PGx-guided n = 52, TAU n = 48) | PGx-Guided vs. TAU:
|
|
| AB-GEN study [23] |
| Spain | 280 (PGx-guided n = 136, TAU n = 144) | PGx-Guided vs. TAU:
|
|
| GENEPSI study [25] |
| Spain | 70 (PGx-guided n = 38, unguided n = 32) | PGx-Guided vs. unguided:
|
|
| Study | Variable | PGx-Guided | Control | p-Value |
|---|---|---|---|---|
| Korean study [24] | CGI-S, mean ± SD | 4.90 ± 0.80 | 4.60 ± 0.70 | 0.063 |
| HDRS-17, mean ± SD | 24.50 ± 4.60 | 23.10 ± 5.00 | 0.159 | |
| AB-GEN study [23] | CGI-S, mean ± SD | 4.50 ± 0.62 | 4.40 ± 0.57 | 0.166 |
| HDRS-17, mean ± SD | 19.47 ± 5.96 | 19.01 ± 5.71 | 0.482 | |
| GENEPSI study [25] | CGI-S, mean ± SD | 4.29 ± 0.57 | 4.26 ± 0.72 | 0.836 |
| HDRS-17, mean ± SD | na | na | na |
| Bias | Korea Study [24] | AB-GEN Study [23] |
|---|---|---|
| Sequence generation (selection bias) | Low: | Low: |
| “Randomization was stratified by study center with a 1:1 ratio for PGx and TAU group, with the use of a random list generated by a computer” | “Randomization was stratified by center with a 1:1 ratio for intervention and control group, using a computer-generated random list” | |
| Location concealment (selection bias) | Low: | Low: |
| Randomization list created at an independent center | Randomization list created at an independent center | |
| Blinding of participants and researchers (performance bias) | High: | High: |
| Patients blinded | Patients blinded | |
| Treating clinician unblinded | Treating clinician unblinded | |
| Blinding of outcome assessment (detection bias) | High: | High: |
| CGI-S and HDRS-17 evaluated by the unblinded treating clinician | CGI-S and HDRS-17 evaluated by the unblinded treating clinician | |
| Incomplete outcome data (attrition bias) | Low: | Low: |
| Patients lost to follow-up were evenly distributed | Patients lost to follow-up were evenly distributed | |
| Selective reporting (reporting bias) | Low: | Low: |
| Prespecified outcomes were reported | Prespecified outcomes were reported | |
| Other sources of bias | High: | High: |
| Patients recruited by the treating clinician | Patients recruited by the treating clinician | |
| Non-industry sponsored | Industry sponsored |
| Bias | GENEPSI Study [25] |
|---|---|
| Confounding (allocation bias) | Low: |
| “Patient data were retrospectively pooled from three psychiatric clinics in Madrid (see author affiliations) that had been using the Neuropharmagen test” | |
| Balanced distribution regarding baseline severity (CGI-S), age, sex, substance abuse and concomitant diseases | |
| “Genetic testing could also result in increased placebo effect. In order to avoid these confounder effects, we sought to perform this retrospective study exclusively in patients who had been genotyped, rather than comparing patients who received pharmacogenetic testing to patients treated as usual.” | |
| Selection of participants (Inception bias) | Low: |
| Baseline visit established as the one in which the saliva sample from the patient was collected | |
| Follow-up visit established 12-weeks after the baseline visit | |
| Misclassification of interventions (misclassification bias) | Moderate: |
| Intervention status well defined, although determined retrospectively | |
| Deviations from intended interventions (performance bias) | Low: |
| Retrospective assignment of intervention. No differences between groups in the care provided | |
| Missing data (attrition bias) | Low: |
| Patients lost to follow-up were evenly distributed | |
| Measurement of outcomes (detection bias) | Moderate: |
| CGI-S recorded in the clinical history by the treating clinician prior to retrospective study protocol definition | |
| Selective reporting (outcome reporting bias) | Low: |
| Prespecified outcomes were reported |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilches, S.; Tuson, M.; Vieta, E.; Álvarez, E.; Espadaler, J. Effectiveness of a Pharmacogenetic Tool at Improving Treatment Efficacy in Major Depressive Disorder: A Meta-Analysis of Three Clinical Studies. Pharmaceutics 2019, 11, 453. https://doi.org/10.3390/pharmaceutics11090453
Vilches S, Tuson M, Vieta E, Álvarez E, Espadaler J. Effectiveness of a Pharmacogenetic Tool at Improving Treatment Efficacy in Major Depressive Disorder: A Meta-Analysis of Three Clinical Studies. Pharmaceutics. 2019; 11(9):453. https://doi.org/10.3390/pharmaceutics11090453
Chicago/Turabian StyleVilches, Silvia, Miquel Tuson, Eduard Vieta, Enric Álvarez, and Jordi Espadaler. 2019. "Effectiveness of a Pharmacogenetic Tool at Improving Treatment Efficacy in Major Depressive Disorder: A Meta-Analysis of Three Clinical Studies" Pharmaceutics 11, no. 9: 453. https://doi.org/10.3390/pharmaceutics11090453