Effect of Ticagrelor, a Cytochrome P450 3A4 Inhibitor, on the Pharmacokinetics of Tadalafil in Rats

 , , and
, , and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. LC-MS/MS Analysis of Tadalafil
2.3. Animals
2.4. Pharmacokinetic Study
2.5. Sample Preparation for LC-MS/MS Analysis
2.6. Pharmacokinetic Data Analysis
2.7. Pharmacokinetic Modeling
2.8. Statistical Analysis
3. Results and Discussion
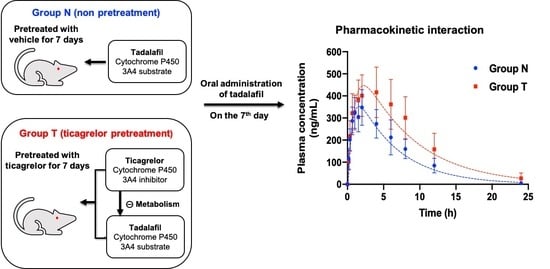
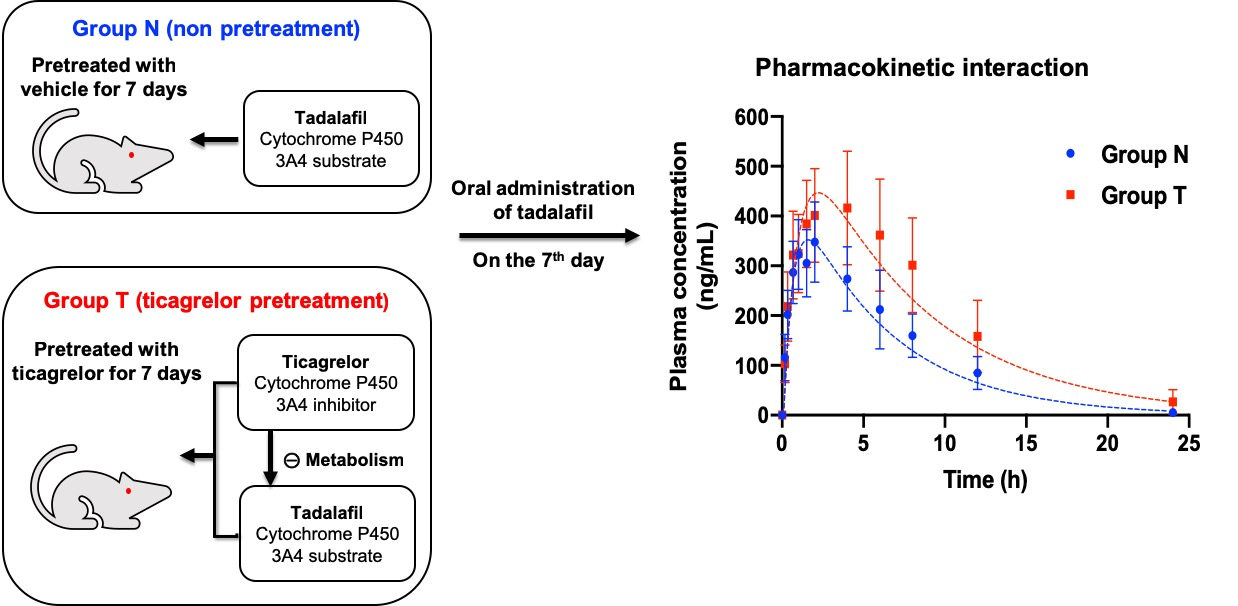
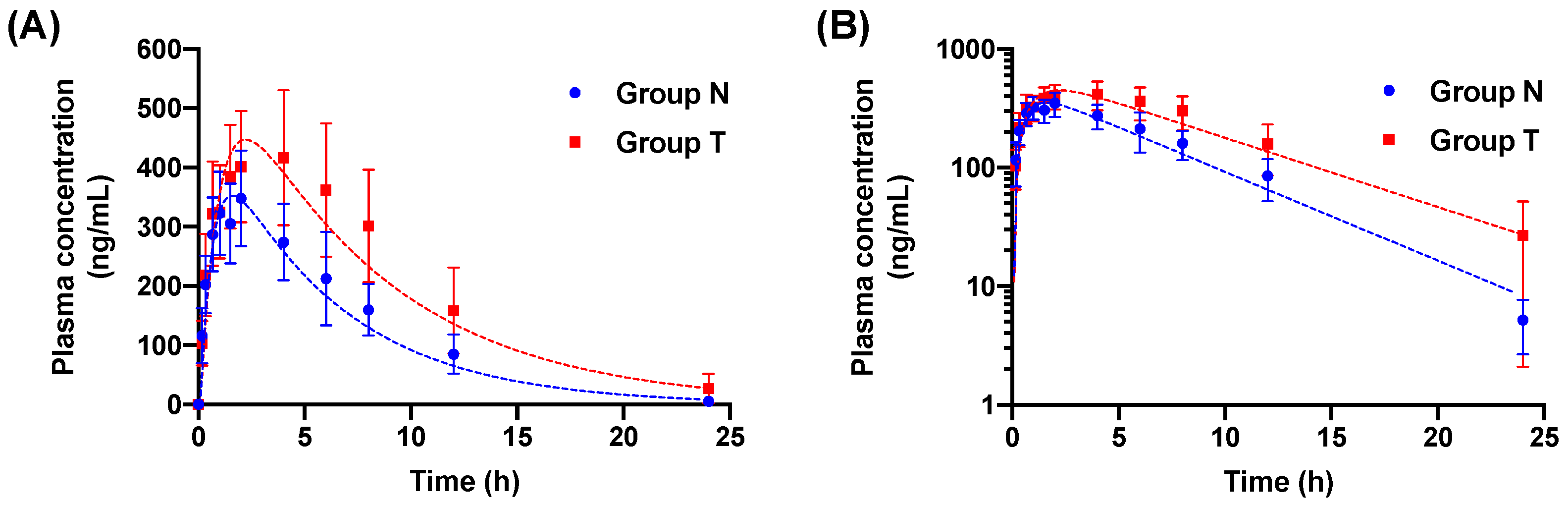
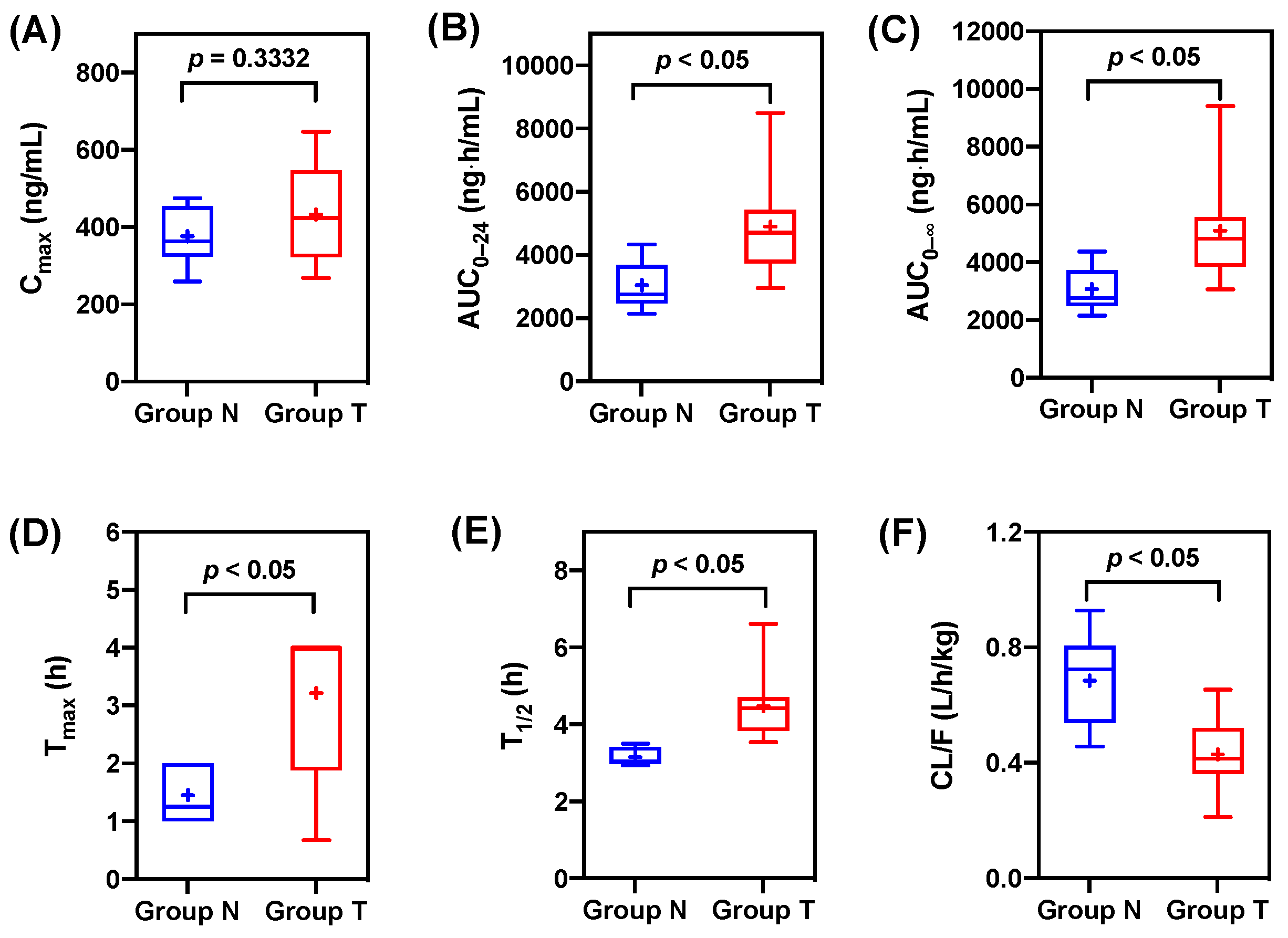
3.1. Pharmacokinetic Data
3.2. Pharmacokinetic Modeling
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hatzimouratidis, K.; Amar, E.; Eardley, I.; Giuliano, F.; Hatzichristou, D.; Montorsi, F.; Vardi, Y.; Wespes, E. Guidelines on male sexual dysfunction: Erectile dysfunction and premature ejaculation. Eur. Urol. 2010, 57, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Laumann, E.O.; Nicolosi, A.; Glasser, D.B.; Paik, A.; Gingell, C.; Moreira, E.; Wang, T. Sexual problems among women and men aged 40–80 y: Prevalence and correlates identified in the global study of sexual attitudes and behaviors. Int. J. Import. Res. 2005, 17, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Dégano, I.R.; Marrugat, J.; Grau, M.; Salvador-González, B.; Ramos, R.; Zamora, A.; Martí, R.; Elosua, R. The association between education and cardiovascular disease incidence is mediated by hypertension, diabetes, and body mass index. Sci. Rep. 2017, 7, 12370. [Google Scholar] [CrossRef] [PubMed]
- Gandaglia, G.; Briganti, A.; Jackson, G.; Kloner, R.A.; Montorsi, F.; Montorsi, P.; Vlachopoulos, C. A systematic review of the association between erectile dysfunction and cardiovascular disease. Eur. Urol. 2014, 65, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Montorsi, P.; Ravagnani, P.M.; Galli, S.; Rotatori, F.; Briganti, A.; Salonia, A.; Dehò, F.; Montorsi, F. Common grounds for erectile dysfunction and coronary artery disease. Curr. Opin. Urol. 2004, 14, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.; Boon, N.; Eardley, I.; Kirby, M.; Dean, J.; Hackett, G.; Montorsi, P.; Montorsi, F.; Vlachopoulos, C.; Kloner, R.; et al. Erectile dysfunction and coronary artery disease prediction: Evidence-based guidance and consensus. Int. J. Clin. Pract. 2010, 64, 848–857. [Google Scholar] [CrossRef]
- Schwarz, E.R.; Rastogi, S.; Kapur, V.; Sulemanjee, N.; Rodriguez, J.J. Erectile dysfunction in heart failure patients. J. Am. Coll. Cardiol. 2006, 48, 1111–1119. [Google Scholar] [CrossRef]
- Speel, T.G.; Van Langen, H.; Meuleman, E.J. The risk of coronary heart disease in men with erectile dysfunction. Eur. Urol. 2003, 44, 366–371. [Google Scholar] [CrossRef]
- Montorsi, P.; Ravagnani, P.M.; Galli, S.; Rotatori, F.; Veglia, F.; Briganti, A.; Salonia, A.; Deho, F.; Rigatti, P.; Montorsi, F.; et al. Association between erectile dysfunction and coronary artery disease. Role of coronary clinical presentation and extent of coronary vessels involvement: The COBRA trial. Eur. Heart J. 2006, 27, 2632–2639. [Google Scholar] [CrossRef]
- Kloner, R.A.; Mullin, S.H.; Shook, T.; Matthews, R.; Mayeda, G.; Burstein, S.; Peled, H.; Pollick, C.; Choudhary, R.; Rosen, R.; et al. Erectile dysfunction in the cardiac patient: How common and should we treat? J. Urol. 2003, 170, S46–S50. [Google Scholar] [CrossRef]
- Nascimento, E.R.; Maia, A.C.; Pereira, V.; Soares-Filho, G.; Nardi, A.E.; Silva, A.C. Sexual dysfunction and cardiovascular diseases: A systematic review of prevalence. Clinics 2013, 68, 1462–1468. [Google Scholar] [CrossRef]
- Coward, R.M.; Carson, C.C. Tadalafil in the treatment of erectile dysfunction. Ther. Clin. Risk Manag. 2008, 4, 1315–1330. [Google Scholar] [CrossRef]
- Clappers, N.; Brouwer, M.A.; Verheugt, F.W. Antiplatelet treatment for coronary heart disease. Heart 2007, 93, 258–265. [Google Scholar] [CrossRef]
- Huang, S.A.; Lie, J.D. Phosphodiesterase-5 (PDE5) inhibitors in the management of erectile dysfunction. Pharm. Ther. 2013, 38, 407–419. [Google Scholar]
- Gong, B.; Ma, M.; Xie, W.; Yang, X.; Huang, Y.; Sun, T.; Luo, Y.; Huang, J. Direct comparison of tadalafil with sildenafil for the treatment of erectile dysfunction: A systematic review and meta-analysis. Int. Urol. Nephrol. 2017, 49, 1731–1740. [Google Scholar] [CrossRef]
- Blount, M.A.; Beasley, A.; Zoraghi, R.; Sekhar, K.R.; Bessay, E.P.; Francis, S.H.; Corbin, J.D. Binding of tritiated sildenafil, tadalafil, or vardenafil to the phosphodiesterase-5 catalytic site displays potency, specificity, heterogeneity, and cGMP stimulation. Mol. Pharmacol. 2004, 66, 144–152. [Google Scholar] [CrossRef]
- Rezvanfar, M.A.; Rahimi, H.R.; Abdollahi, M. ADMET considerations for phosphodiesterase-5 inhibitors. Expert Opin. Drug Metab. Toxicol. 2012, 8, 1231–1245. [Google Scholar] [CrossRef]
- Na, Y.G.; Byeon, J.J.; Wang, M.; Huh, H.W.; Son, G.H.; Jeon, S.H.; Bang, K.H.; Kim, S.J.; Lee, H.J.; Lee, H.K.; et al. Strategic approach to developing a self-microemulsifying drug delivery system to enhance antiplatelet activity and bioavailability of ticagrelor. Int. J. Nanomed. 2019, 14, 1193–1212. [Google Scholar] [CrossRef]
- Son, G.H.; Na, Y.G.; Huh, H.W.; Wang, M.; Kim, M.K.; Han, M.G.; Byeon, J.J.; Lee, H.K.; Cho, C.W. Systemic design and evaluation of ticagrelor-loaded nanostructured lipid carriers for enhancing bioavailability and antiplatelet activity. Pharmaceutics 2019, 11, E222. [Google Scholar] [CrossRef]
- Bliden, K.P.; Tantry, U.S.; Storey, R.F.; Jeong, Y.H.; Gesheff, M.; Wei, C.; Gurbel, P.A. The effect of ticagrelor versus clopidogrel on high on-treatment platelet reactivity: Combined analysis of the ONSET/OFFSET and RESPOND studies. Am. Heart J. 2011, 162, 160–165. [Google Scholar] [CrossRef]
- Teng, R.; Butler, K. The effect of ticagrelor on the metabolism of midazolam in healthy volunteers. Clin. Ther. 2013, 35, 1025–1037. [Google Scholar] [CrossRef]
- Zhou, D.; Andersson, T.B.; Grimm, S.W. In vitro evaluation of potential drug-drug interactions with ticagrelor: Cytochrome P450 reaction phenotyping, inhibition, induction, and differential kinetics. Drug Metab. Dispos. 2011, 39, 703–710. [Google Scholar] [CrossRef]
- Teng, R.; Mitchell, P.D.; Butler, K.A. Pharmacokinetic interaction studies of co-administration of ticagrelor and atorvastatin or simvastatin in healthy volunteers. Eur. J. Clin. Pharmacol. 2013, 69, 477–487. [Google Scholar] [CrossRef]
- Klein, K.; Zanger, U.M. Pharmacogenomics of cytochrome P450 3A4: Recent progress toward the “missing heritability” problem. Front. Genet. 2013, 4, 1–12. [Google Scholar] [CrossRef]
- Garraffo, R.; Lavrut, T.; Ferrando, S.; Durant, J.; Rouyrre, N.; MacGregor, T.R.; Sabo, J.P.; Dellamonica, P. Effect of tipranavir/ritonavir combination on the pharmacokinetics of tadalafil in healthy volunteers. J. Clin. Pharmacol. 2011, 51, 1071–1078. [Google Scholar] [CrossRef]
- Fraunfelder, F.W. Visual side effects associated with erectile dysfunction agents. Am. J. Oppthalmol. 2005, 140, 723–724. [Google Scholar] [CrossRef]
- Montorsi, F.; Verheyden, B.; Meuleman, E.; Jünemann, K.P.; Moncada, I.; Valiquette, L.; Casabe, A.; Pacheco, C.; Denne, J.; Knight, J.; et al. Long-term safety and tolerability of tadalafil in the treatment of erectile dysfunction. Eur. Urol. 2004, 45, 339–345. [Google Scholar] [CrossRef]
- Porst, H.; Giuliano, F.; Glina, S.; Ralph, D.; Casabé, A.R.; Elion-Mboussa, A.; Shen, W.; Whitaker, J.S. Evaluation of the efficacy and safety of once-a-day dosing of tadalafil 5 mg and 10 mg in the treatment of erectile dysfunction: Results of a multicenter, randomized, double-blind, placebo-controlled trial. Eur. Urol. 2006, 50, 351–359. [Google Scholar] [CrossRef]
- Hatzichristou, D.; Gambla, M.; Rubio-Aurioles, E.; Buvat, J.; Brock, G.B.; Spera, G.; Rose, L.; Lording, D.; Liang, S. Efficacy of tadalafil once daily in men with diabetes mellitus and erectile dysfunction. Diabet. Med. 2008, 25, 138–146. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R. Bioanalytical method validation: An updated review. Pharm. Methods 2010, 1, 25–38. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Guidance for Industry and Reviewers: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. 2005. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/estimating-maximum-safe-starting-dose-initial-clinical-trials-therapeutics-adult-healthy-volunteers (accessed on 24 August 2018).
- Bang, K.H.; Na, Y.G.; Huh, H.W.; Hwang, S.J.; Kim, M.S.; Kim, M.; Lee, H.K.; Cho, C.W. The delivery strategy of paclitaxel nanostructured lipid carrier coated with platelet membrane. Cancers 2019, 11, 807. [Google Scholar] [CrossRef]
- Kim, M.S.; Baek, I.H. Effect of dronedarone on the pharmacokinetics of carvedilol following oral administration to rats. Eur. J. Pharm. Sci. 2018, 111, 13–19. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, M.S.; Baek, I.H. Enhanced bioavailability of tadalafil after intranasal administration in beagle dogs. Pharmaceutics 2018, 10, 187. [Google Scholar] [CrossRef]
- Sjögren, E.; Thorn, H.; Tannergren, C. In silico modeling of gastrointestinal drug absorption: Predictive performance of three physiologically based absorption models. Mol. Pharm. 2016, 13, 1763–1778. [Google Scholar] [CrossRef]
- Teng, R. Ticagrelor: Pharmacokinetic, pharmacodynamic and pharmacogenetic profile: An update. Clin. Pharmacokinet. 2015, 54, 1125–1138. [Google Scholar] [CrossRef]
- Kothare, P.A.; Seger, M.E.; Northrup, J.; Mace, K.; Mitchell, M.I.; Linnebjerg, H. Effect of exenatide on the pharmacokinetics of a combination oral contraceptive in healthy women: An open-label, randomised, crossover trial. BMC Clin. Pharmacol. 2012, 12, 8. [Google Scholar] [CrossRef]
- Doligalski, C.T.; Tong Logan, A.; Silverman, A. Drug interactions: A primer for the gastroenterologist. Gastroenterol. Hepatol. 2012, 8, 376–383. [Google Scholar]
- Jetter, A.; Kinzig-Schippers, M.; Walchner-Bonjean, M.; Hering, U.; Bulitta, J.; Schreiner, P.; Sörgel, F.; Fuhr, U. Effects of grapefruit juice on the pharmacokinetics of sildenafil. Clin. Pharmacol. Ther. 2002, 71, 21–29. [Google Scholar] [CrossRef]
- Lee, D.S.; Kim, S.J.; Choi, G.W.; Lee, Y.B.; Cho, H.Y. Pharmacokinetic–pharmacodynamic model for the testosterone-suppressive effect of leuprolide in normal and prostate cancer rats. Molecules 2018, 23, 909. [Google Scholar] [CrossRef]
- Wagner, C.; Pan, Y.; Hsu, V.; Grillo, J.A.; Zhang, L.; Reynolds, K.S.; Sinha, V.; Zhao, P. Predicting the effect of cytochrome P450 inhibitors on substrate drugs: Analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin. Pharmacokinet. 2015, 54, 117–127. [Google Scholar] [CrossRef]
- Teng, R.; Butler, K. Effect of the CYP3A inhibitors, diltiazem and ketoconazole, on ticagrelor pharmacokinetics in healthy volunteers. J. Drug Assess. 2013, 2, 30–39. [Google Scholar] [CrossRef]
- Fransis, S.H.; Corbin, J.D. Molecular mechanisms and pharmacokinetics of phosphodiesterase-5 antagonists. Curr. Urol. Rep. 2003, 4, 457–465. [Google Scholar] [CrossRef]
- Kloner, R.A.; Hutter, A.M.; Emmick, J.T.; Mitchell, M.I.; Denne, J.; Jackson, G. Time course of the interaction between tadalafil and nitrates. J. Am. Coll. Cardiol. 2003, 42, 1855–1860. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
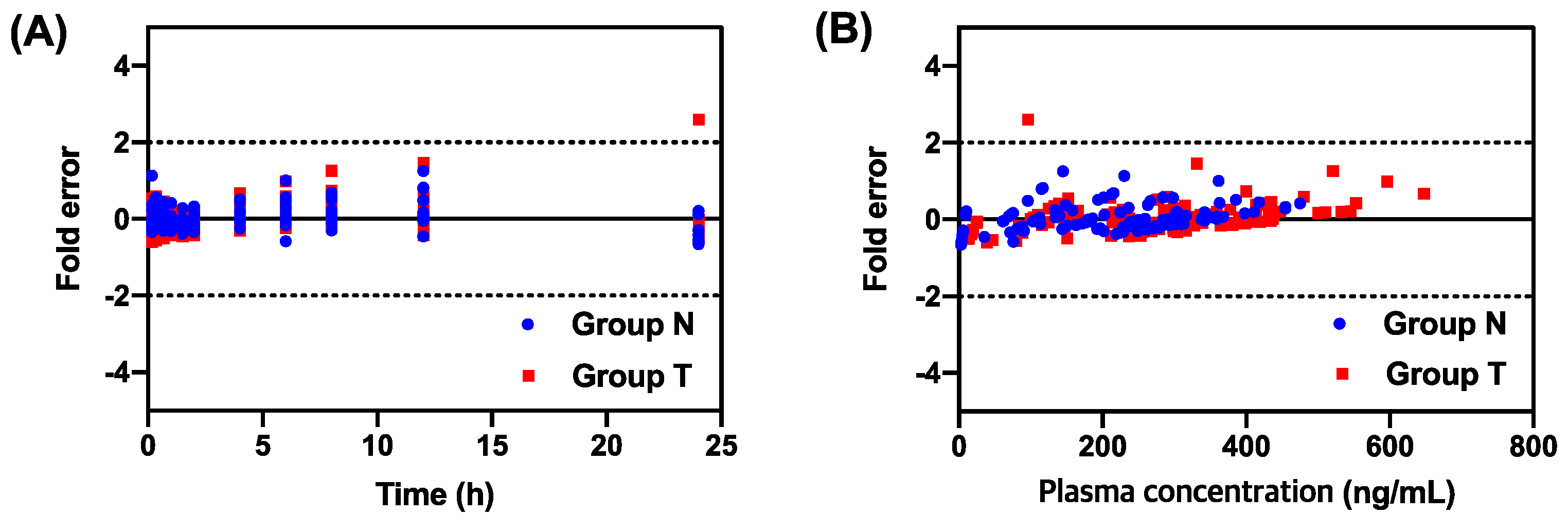{kind=link}
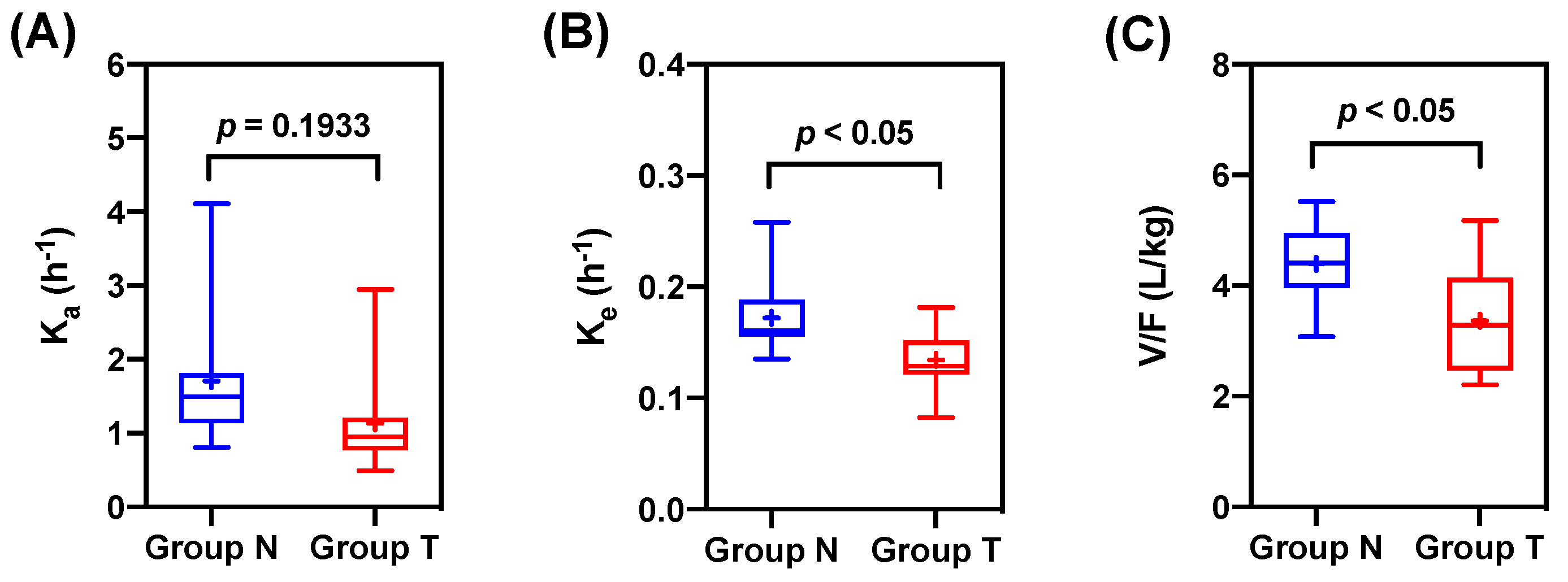{kind=link}
| Parameters | Group N | Group T | Ratio a | p-Value |
|---|---|---|---|---|
| Cmax (ng/mL) | 375.94 ± 72.81 | 432.71 ± 119.79 | 1.15 | 0.3332 |
| AUC0–24 (ng⋅h/mL) | 3046.88 ± 732.50 | 4895.74 ± 1592.87 | 1.61 | 0.0124 |
| AUC0–∞ (ng⋅h/mL) | 3070.95 ± 743.01 | 5095.04 ± 1800.30 | 1.66 | 0.0134 |
| Tmax (h) | 1.45 ± 0.50 | 3.22 ± 1.30 | 2.22 | 0.0030 |
| T1/2 (h) | 3.15 ± 0.23 | 4.47 ± 0.89 | 1.42 | 0.0018 |
| CL/F (L/h/kg) | 0.68 ± 0.15 | 0.43 ± 0.13 | 0.63 | 0.0035 |
| Parameters | Group N | Group T | Ratio a | p-Value |
|---|---|---|---|---|
| Ka (1/h) | 1.71 ± 0.91 | 1.14 ± 0.73 | 0.67 | 0.1933 |
| Ke (1/h) | 0.17 ± 0.03 | 0.13 ± 0.03 | 0.77 | 0.0166 |
| V/F (L/kg) | 4.39 ± 0.76 | 3.36 ± 0.95 | 0.77 | 0.0438 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Na, Y.-G.; Byeon, J.-J.; Huh, H.W.; Kim, M.-K.; Shin, Y.G.; Lee, H.-K.; Cho, C.-W. Effect of Ticagrelor, a Cytochrome P450 3A4 Inhibitor, on the Pharmacokinetics of Tadalafil in Rats. Pharmaceutics 2019, 11, 354. https://doi.org/10.3390/pharmaceutics11070354
Na Y-G, Byeon J-J, Huh HW, Kim M-K, Shin YG, Lee H-K, Cho C-W. Effect of Ticagrelor, a Cytochrome P450 3A4 Inhibitor, on the Pharmacokinetics of Tadalafil in Rats. Pharmaceutics. 2019; 11(7):354. https://doi.org/10.3390/pharmaceutics11070354
Chicago/Turabian StyleNa, Young-Guk, Jin-Ju Byeon, Hyun Wook Huh, Min-Ki Kim, Young G. Shin, Hong-Ki Lee, and Cheong-Weon Cho. 2019. "Effect of Ticagrelor, a Cytochrome P450 3A4 Inhibitor, on the Pharmacokinetics of Tadalafil in Rats" Pharmaceutics 11, no. 7: 354. https://doi.org/10.3390/pharmaceutics11070354