Repurposing Butenafine as An Oral Nanomedicine for Visceral Leishmaniasis


 and
and
Abstract
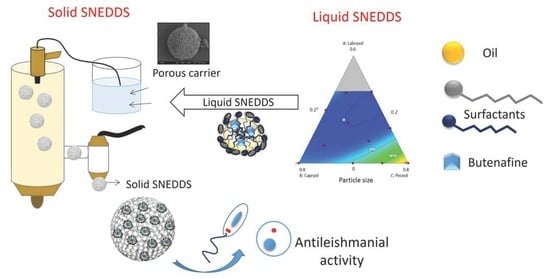
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Solubility Studies of Butenafine
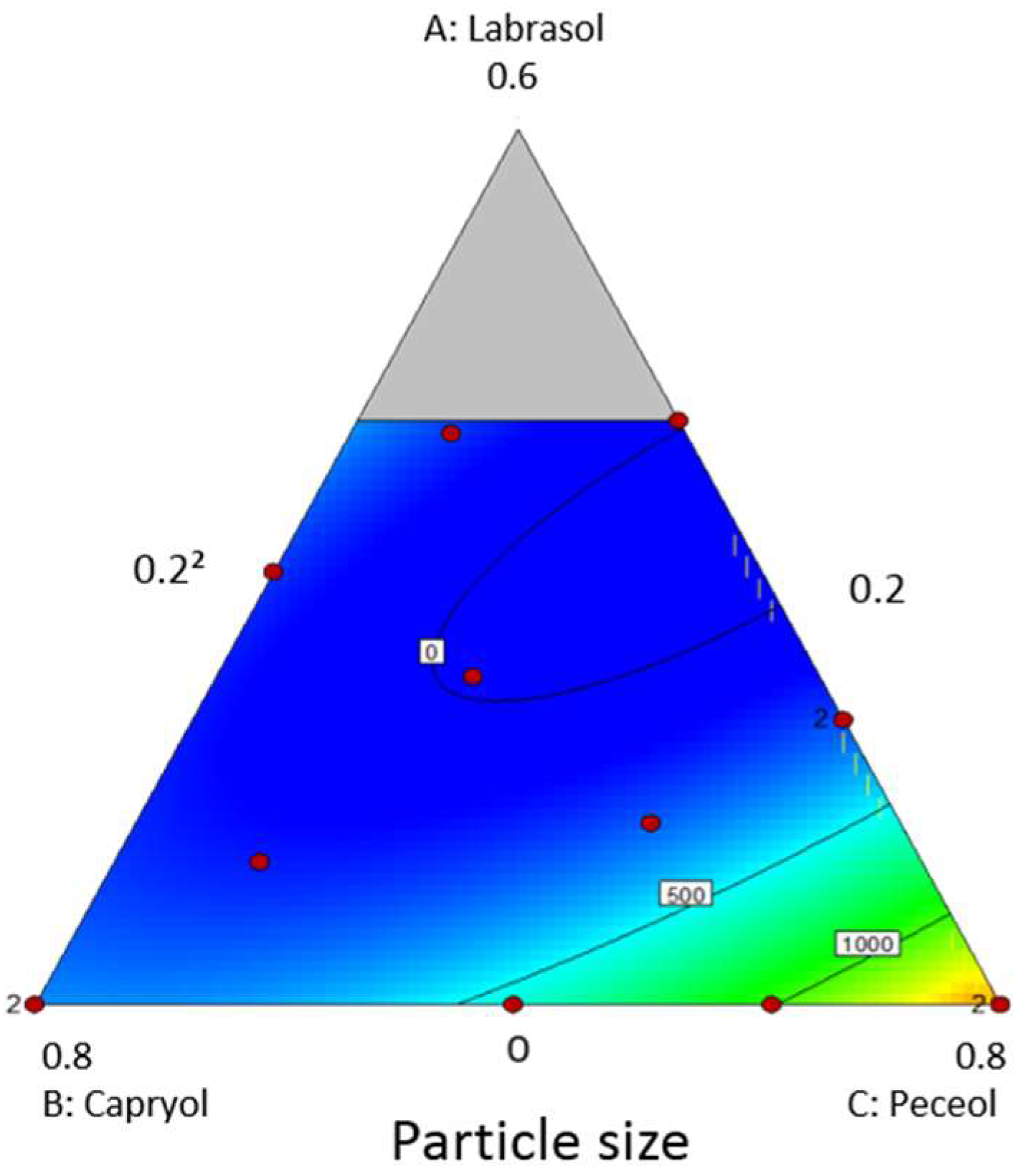
2.3. Pseudo-Ternary Phase Diagrams
2.4. Preparation of Liquid B-SNEDDS Formulations
2.5. Preparation of Solid B-SNEDDS Formulation
2.6. Characterisation of the Solid B-SNEDDS
2.6.1. Powder Flow: Angle of Repose (AoR) Measurements
2.6.2. Particle Size Measurements
2.6.3. Release Studies
2.6.4. Drug Loading
2.6.5. Morphological Analysis
2.6.6. Tabletting and Hardness
2.7. In Vitro Efficacy and Toxicity Studies
2.7.1. Parasites and Cell Lines
2.7.2. In Vitro Promastigote Efficacy and Cytotoxicity
2.7.3. Macrophage Infection and Treatments
2.8. Statistical Analysis
3. Results
3.1. Solubility Studies of Butenafine
3.2. Pseudo-Ternary Phase Diagrams and Preparation of Liquid B-SNEDDS Formulations
- (1)
- Combination A: Peceol:Capryol:Labrasol mixed at 27:58:15 w/w
- (2)
- Combination B: Peceol:Capryol:Labrasol mixed at 25:51:24 w/w
3.3. Yield of Solid B-SNEDDS
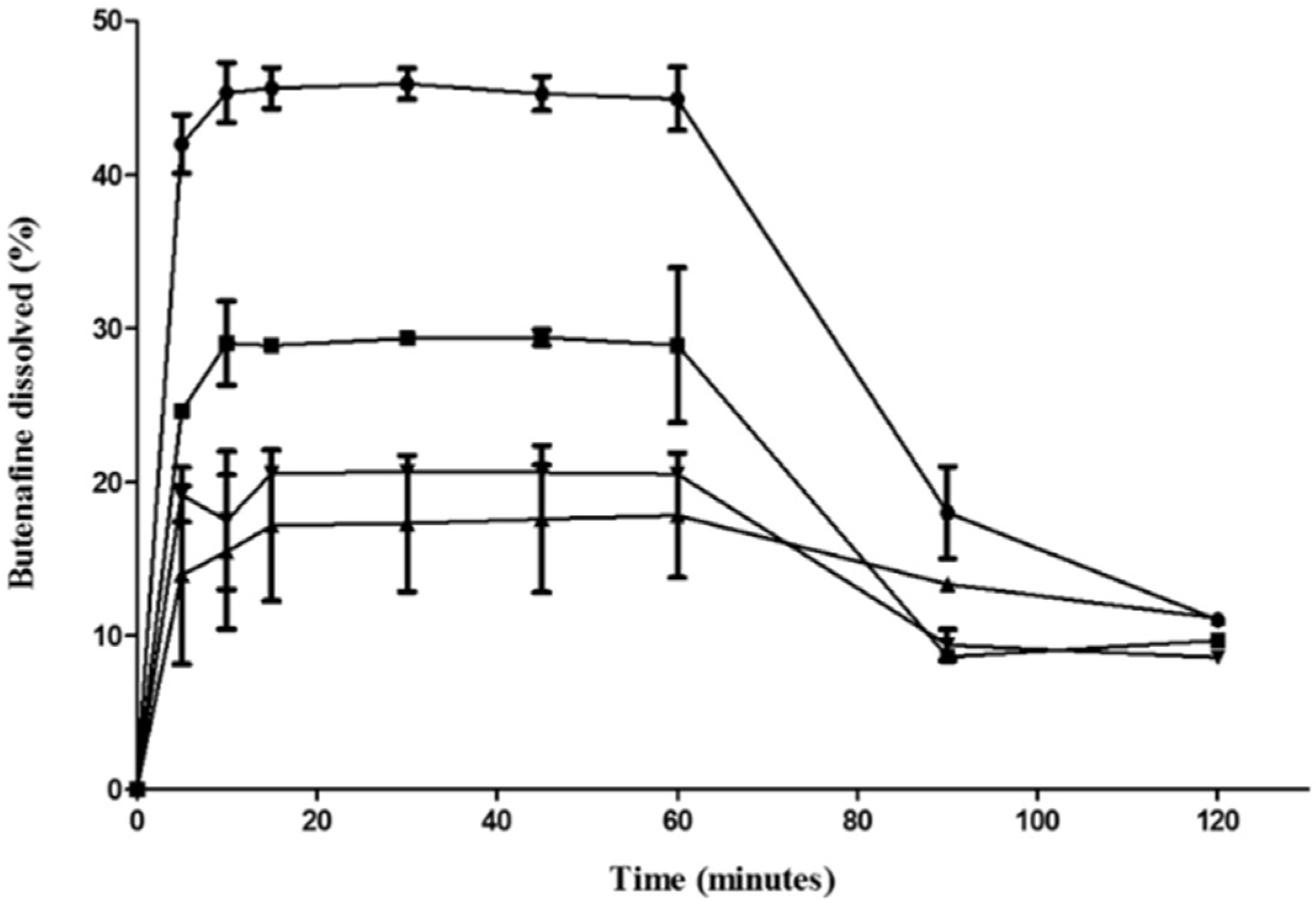
3.4. Release Studies
3.5. Morphological Analysis
3.6. Tableting and Hardness
3.7. In Vitro Efficacy and Cytotoxicity
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Araújo-Santos, T.; Prates, D.B.; Andrade, B.B.; Nascimento, D.O.; Clarêncio, J.; Entringer, P.F.; Carneiro, A.B.; Silva-Neto, M.A.; Miranda, J.C.; Brodskyn, C.I.; et al. Lutzomyia longipalpis saliva triggers lipid body formation and prostaglandin E₂ production in murine macrophages. PLoS Negl. Trop. Dis. 2010, 4, e873. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Leishmaniasis Disease and Epidemiology. Available online: http://www.who.int/leishmaniasis/epidemic/response_more/en/index.html (accessed on 18 February 2019).
- Rittig, M.G.; Bogdan, C. Leishmania-Host-cell Interaction: Complexities and Alternative Views. Parasitol. Today 2000, 16, 292–297. [Google Scholar] [CrossRef]
- Laurenti, M.D.; Da Matta, V.L.R.; Pernichelli, T.; Secundino, N.F.C.; Pinto, L.C.; Corbett, C.E.P.; Pimenta, P.P.F. Effects of Salivary Gland Homogenate from Wild-Caught and Laboratory-Reared Lutzomyia longipalpison the Evolution and Immunomodulation of Leishmania (Leishmania) amazonensis Infection. Scand. J. Immunol. 2009, 70, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Laison, R.; Shaw, J.J. New world Leishmaniasis—The Neotropical Leishmania species. In Topley & Wilson. Microbiology and Microbial Infections, 9th ed.; Feg Cox: London, UK, 1988. [Google Scholar]
- Silveira, F.T.; Lainson, R.; Crescente, J.Â.; De Souza, A.A.; Campos, M.B.; Gomes, C.M.; Laurenti, M.D.; Corbett, C.E. A prospective study on the dynamics of the clinical and immunological evolution of human Leishmania (L.) infantum chagasi infection in the Brazilian Amazon region. Trans. R. Soc. Trop. Med. Hyg. 2010, 104, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Dantas-Torres, F. Leishmania infantum versus Leishmania chagasi: Do not forget the law of priority. Memórias do Instituto Oswaldo Cruz 2006, 101, 117–118. [Google Scholar] [CrossRef]
- Barak, E.; Amin-Spector, S.; Gerliak, E.; Goyard, S.; Holland, N.; Zilberstein, D. Differentiation of Leishmania donovani in host-free system: Analysis of signal perception and response. Mol. Biochem. Parasitol. 2005, 141, 99–108. [Google Scholar] [CrossRef]
- Santos, D.O.; Coutinho, C.E.R.; Madeira, M.F.; Bottino, C.G.; Vieira, R.T.; Nascimento, S.B.; Bernardino, A.M.R.; Bourguignon, S.C.; Côrte-Real, S.; Pinho, R.T.; et al. Leishmaniasis treatment—A challenge that remains: A review. Parasitol. Res. 2008, 103, 1–10. [Google Scholar] [CrossRef]
- Kaur, G.; Rajput, B. Comparative Analysis of the Omics Technologies Used to Study Antimonial, Amphotericin B, and Pentamidine Resistance in Leishmania. J. Parasitol. Res. 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Serrano, D.R.; Lalatsa, A. Oral amphotericin B: The journey from bench to market. J. Drug Deliv. Sci. Technol. 2017, 42, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.; Serrano, D.R.; Mauger, M.; Bolás-Fernández, F.; Dea-Ayuela, M.A.; Lalatsa, A. Orally Bioavailable and Effective Buparvaquone Lipid-Based Nanomedicines for Visceral Leishmaniasis. Mol. Pharm. 2018, 15, 2570–2583. [Google Scholar] [CrossRef]
- Fernández, O.L.; Díaz-Toro, Y.; Ovalle, C.; Valderrama, L.; Muvdi, S.; Rodriguez, I.; Gomez, M.A.; Saravia, N.G. Miltefosine and Antimonial Drug Susceptibility of Leishmania Viannia Species and Populations in Regions of High Transmission in Colombia. PLoS Negl. Trop. Dis. 2014, 8, e2871. [Google Scholar] [CrossRef]
- Dorlo, T.P.C.; Balasegaram, M.; Beijnen, J.H.; De Vries, P.J. Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2576–2597. [Google Scholar] [CrossRef]
- Pérez-Victoria, F.J.; Sánchez-Cañete, M.P.; Seifert, K.; Croft, S.L.; Sundar, S.; Castanys, S.; Gamarro, F. Mechanisms of experimental resistance of Leishmania to miltefosine: Implications for clinical use. Drug Resist. Updates 2006, 9, 26–39. [Google Scholar] [CrossRef]
- Drugs for Neglected Diseases initiative: DNDi. Available online: https://www.dndi.org/diseases-projects/leishmaniasis/tpp-vl (accessed on 19 January 2018).
- Graul, A.; Sorbera, L.; Pina, P.; Tell, M.; Cruces, E.; Rosa, E.; Stringer, M.; Castaner, R.; Revel, L. The year’s new drugs & biologics–2009. Drug News Perspect. 2010, 23, 7. [Google Scholar] [CrossRef]
- Tobinick, E.L. The value of drug repositioning in the current pharmaceutical market. Drug News Perspect. 2009, 22, 119. [Google Scholar] [CrossRef]
- Voss, A.; Soto, J.; Toledo, J.; Nicholls, R.S.; Padilla, J.; Engel, J.; Fischer, C.; Gutierrez, P.; Berman, J. Treatment of American Cutaneous Leishmaniasis with Miltefosine, an Oral Agent. Clin. Infect. Dis. 2001, 33, e57–e61. [Google Scholar] [Green Version]
- Bezerra-Souza, A.; Yamamoto, E.S.; Laurenti, M.D.; Ribeiro, S.P.; Passero, L.F.D. The antifungal compound 25butenafine eliminates promastigote and amastigote forms of Leishmania (Leishmania) amazonensis and Leishmania (Viannia) braziliensis. Parasitol. Int. 2016, 65, 702–707. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA). Review and Evaluation of Pharmacology/Toxicology Data O Fbutenafine, NDA 21-307 (000)/09-29-2000. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/21-307_Lotrimin_pharmr.pdf (accessed on 24 June 2019).
- Serrano, D.R.; Lalatsa, A.; Dea-Ayuela, M.A. Engineering Synergistically Active and Bioavailable Cost-effective Medicines for Neglected Tropical Diseases; The Role of Excipients. Curr. Top. Med. Chem. 2017, 17. [Google Scholar] [CrossRef]
- Lavoie, F.; Cartilier, L.; Thibert, R. New Methods Characterizing Avalanche Behavior to Determine Powder Flow. Pharm. Res. 2002, 19, 887–893. [Google Scholar] [CrossRef]
- Betatek Inc. Instrumentation Superbly Supported: Microtrac Zetatrac Nanotechnology, Particle Size and Charge Measurement Analyzer; Betatek Inc.: Toronto, ON, Canada, 2012. [Google Scholar]
- United States Pharmacopeia (USP). USP 41-NF36; <711> Dissolution Studies; United States Pharmacopeial Convention: Rockville, MD, USA, 2008. [Google Scholar]
- Drug Bank Database. Available online: https://www.drugbank.ca/drugs/DB01091 (accessed on 15 July 2019).
- European Pharmacopeia, 9th ed. Available online: https://www.edqm.eu/en/european-pharmacopoeia-ph-eur-9th-edition. (accessed on 19 July 2018).
- Passero, L.F.D.; Sacomori, J.V.; Tomokane, T.Y.; Corbett, C.E.P.; Da Silveira, F.T.; Laurenti, M.D. Ex vivo and in vivo biological behavior of Leishmania (Viannia) shawi. Parasitol. Res. 2009, 105, 1741–1747. [Google Scholar] [CrossRef]
- Gattefossé: Excipients for Solubility and Bioavailability Enhancement. Available online: https://www.gattefosse.com/excipients-for-solubility-and-bioavailability-enhancement (accessed on 18 February 2019).
- Maher, S.; Brayden, D.J.; Casettari, L.; Illum, L. Application of Permeation Enhancers in Oral Delivery of Macromolecules: An Update. Pharmaceutics 2019, 11, 41. [Google Scholar] [CrossRef]
- United States Pharmacopeia (USP). USP 41-NF36; <1174> Powder Flow; United States Pharmacopeial Convention: Rockville, MD, USA, 2006. [Google Scholar]
- Goh, H.P.; Heng, P.W.S.; Liew, C.V. Comparative evaluation of powder flow parameters with reference to particle size and shape. Int. J. Pharm. 2018, 547, 133–141. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Pouton, C.W.; Cuine, J.F.; Charman, W.N. Enhancing intestinal drug solubilization using lipid-based delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 673–691. [Google Scholar] [CrossRef]
- Humberstone, A.J.; Charman, W.N. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv. Drug Deliv. Rev. 1997, 25, 103–128. [Google Scholar] [CrossRef]
- Drugbank. Butenafine. Available online: https://www.drugbank.ca/drugs/DB01091 (accessed on 29 April 2019).
- Seifert, K.; Escobar, P.; Croft, S.L. In vitro activity of anti-leishmanial drugs against Leishmania donovani is host cell dependent. J. Antimicrob. Chemother. 2010, 65, 508–511. [Google Scholar] [CrossRef] [Green Version]
- Stütz, A. Synthesis and Structure—Activity Correlations within Allylamine Antimycotics. Ann. N. Y. Acad. Sci. 1988, 544, 46–62. [Google Scholar] [CrossRef]
- Ryder, N.S. Terbinafine: Mode of action and properties of the squalene epoxidase inhibition. Br. J. Dermatol. 1992, 126, 2–7. [Google Scholar] [CrossRef]
- Roberts, C.W.; McLeod, R.; Rice, D.W.; Ginger, M.; Chance, M.L.; Goad, L.J. Fatty acid and sterol metabolism: Potential antimicrobial targets in apicomplexan and trypanosomatid parasitic protozoa. Mol. Biochem. Parasitol. 2003, 126, 129–142. [Google Scholar] [CrossRef]
- Singal, A. Butenafine and superficial mycoses: Current status. Expert Opin. Drug Metab. Toxicol. 2008, 4, 999–1005. [Google Scholar] [CrossRef]
- Tatham, L.M.; Rannard, S.P.; Owen, A. Nanoformulation strategies for the enhanced oral bioavailability of antiretroviral therapeutics. Ther. Deliv. 2015, 6, 469–490. [Google Scholar] [CrossRef]
- Anton, N.; Vandamme, T.F. Nano-emulsions and Micro-emulsions: Clarifications of the Critical Differences. Pharm. Res. 2011, 28, 978–985. [Google Scholar] [CrossRef]
- Leonaviciute, G.; Adamovic, N.T.; Lam, H.T.; Rohrer, J.; Partenhauser, A.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems (SEDDS): Proof-of-concept how to make them mucoadhesive. Eur. J. Pharm. Biopharm. 2017, 112, 51–57. [Google Scholar] [CrossRef]
- Imada, C.; Takahashi, T.; Kuramoto, M.; Masuda, K.; Ogawara, K.; Sato, A.; Wataya, Y.; Kim, H.S.; Higaki, K. Improvement of Oral Bioavailability of N-251, a Novel Antimalarial Drug, by Increasing Lymphatic Transport with Long-Chain Fatty Acid-Based Self-Nanoemulsifying Drug Delivery System. Pharm. Res. 2015, 32, 2595–2608. [Google Scholar] [CrossRef]
- Khan, A.A.; Mudassir, J.; Mohtar, N.; Darwis, Y. Advanced drug delivery to the lymphatic system: Lipid-based nanoformulations. Int. J. Nanomed. 2013, 8, 2733–2744. [Google Scholar] [CrossRef]
- Gosh, S.; Roy, T. Nanoparticulate drug-delivery systems: Lymphatic uptake and its gastrointestinal application. J. Appl. Pharm. Sci. 2014, 4, 123–130. [Google Scholar]
- Elnaggar, Y.S.; El-Massik, M.A.; Abdallah, O.Y. Self-nanoemulsifying drug delivery systems of tamoxifen citrate: Design and optimization. Int. J. Pharm. 2009, 380, 133–141. [Google Scholar] [CrossRef]
- Shen, S.; Wu, Y.; Liu, Y.; Wu, D. High drug-loading nanomedicines: Progress, current status, and prospects. Int. J. Nanomed. 2017, 12, 4085–4109. [Google Scholar] [CrossRef]
- Serrano, D.R.; Hernández, L.; Fleire, L.; González-Alvarez, I.; Montoya, A.; Ballesteros, M.P.; Dea-Ayuela, M.A.; Miró, G.; Bolás-Fernández, F.; Torrado, J.J. Hemolytic and pharmacokinetic studies of liposomal and particulate amphotericin B formulations. Int. J. Pharm. 2013, 447, 38–46. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vehicle | Butenafine (mg/g) | HLB * |
|---|---|---|
| Peceol | 24.68 ± 0.21 | 1 |
| Capryol 90 | 15.79 ± 1.18 | 5 |
| Labrasol | 13.03 ± 3.94 | 12 |
| Labrafil M 1944 CS | 22.59 ± 1.66 | 9 |
| Experiment | Labrasol | Capryol 90 | Peceol | Particle Size (nm) |
|---|---|---|---|---|
| 1 | 0.29 | 0.50 | 0.2 | 116 |
| 2 | 0 | 0.50 | 0.49 | 376 |
| 3 | 0 | 0.8 | 0.2 | 289 |
| 4 | 0.19 | 0.2 | 0.60 | 191 |
| 5 | 0.22 | 0.41 | 0.35 | 156 |
| 6 | 0.29 | 0.50 | 0.2 | 178 |
| 7 | 0.09 | 0.61 | 0.29 | 244 |
| 8 | 0 | 0.2 | 0.8 | 1735 |
| 9 | 0 | 0.50 | 0.49 | 410 |
| 10 | 0.39 | 0.34 | 0.26 | 107 |
| 11 | 0.4 | 0.2 | 0.4 | 101 |
| 12 | 0.12 | 0.35 | 0.52 | 167 |
| 13 | 0 | 0.2 | 0.8 | 1340 |
| 14 | 0 | 0.34 | 0.65 | 1180 |
| 15 | 0 | 0.8 | 0.2 | 352 |
| 16 | 0.19 | 0.2 | 0.60 | 141 |
| Code | Aerosil Type | Carrier: B-SNEDDS Ratio (w/w) | Yield (%) | Angle of Repose (°) | Drug Loading (%) | Particle Size (nm) | Butenafine Released at 60 min in SGF (%) |
|---|---|---|---|---|---|---|---|
| F1 | 200 | 1:2 | 71.1 ± 1.6 | 19.1 ± 5.4 | 94.1 ± 0.1 | 119.8 ± 2.3 | 45 ± 2.0 |
| F2 | 200 | 1:3 | 66.5 ±10.2 | 15.9 ± 3.0 | 96.5 ± 2.4 | 25.9 ± 1.2 | 28 ± 5.0 |
| F3 | R972 | 1:2 | 32.9 ± 1.3 | 20.6 ± 0.2 | 85.5 ± 4.6 | 279 ± 8.7 | 18 ± 4.0 |
| F4 | R972 | 1:3 | 47.4 ± 22.6 | 8.7 ± 1.5 | 97.0 ± 2.1 | 1 * | 20 ± 0.2 |
| Compaction Pressure (kN) | Hardness (N) | Dimension (mm) |
|---|---|---|
| 9806.65 | 15.6–18.4 | 12.91 × 3.17 |
| 19,613.3 | 7.7–20.1 | 12.91 × 3.17 |
| Formulation | EC50 (µM) Promastigote | CC50 (µM) Macrophage | EC50 (µM) Amastigote | SIp | SIa |
|---|---|---|---|---|---|
| F1 | 76.5 ± 3.0 * | 225.8 ± 3.2 | 164.4 ± 3.4 | 2.9 | 1.3 |
| F2 | 93.1 ± 3.0 | 127.2 ± 3.0 | 702.5 ± 3.2 | 1.3 | 0.1 |
| F3 | 101.7 ± 3.2 | ≥300.0 | 86.4 ± 2.9 * | ≥3.0 | ≥3.6 |
| F4 | 73.9 ± 3.2 * | 233.1 ± 3.0 | 27.0 ± 3.4 * | 3.1 | 8.6 |
| Butenafine | 99.8 ± 3.1 | 109.3 ± 3.5 | 118.4 ± 3.6 | 1.0 | 0.9 |
| Miltefosine | 17.9 ± 0.9 | 126.3 ± 3.5 | 13.7 ± 0.7 | 7.0 | 9.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezerra-Souza, A.; Fernandez-Garcia, R.; Rodrigues, G.F.; Bolas-Fernandez, F.; Dalastra Laurenti, M.; Passero, L.F.; Lalatsa, A.; Serrano, D.R. Repurposing Butenafine as An Oral Nanomedicine for Visceral Leishmaniasis. Pharmaceutics 2019, 11, 353. https://doi.org/10.3390/pharmaceutics11070353
Bezerra-Souza A, Fernandez-Garcia R, Rodrigues GF, Bolas-Fernandez F, Dalastra Laurenti M, Passero LF, Lalatsa A, Serrano DR. Repurposing Butenafine as An Oral Nanomedicine for Visceral Leishmaniasis. Pharmaceutics. 2019; 11(7):353. https://doi.org/10.3390/pharmaceutics11070353
Chicago/Turabian StyleBezerra-Souza, Adriana, Raquel Fernandez-Garcia, Gabriela F. Rodrigues, Francisco Bolas-Fernandez, Marcia Dalastra Laurenti, Luiz Felipe Passero, Aikaterini Lalatsa, and Dolores R. Serrano. 2019. "Repurposing Butenafine as An Oral Nanomedicine for Visceral Leishmaniasis" Pharmaceutics 11, no. 7: 353. https://doi.org/10.3390/pharmaceutics11070353