Microwave-Assisted Freeze-Drying of Monoclonal Antibodies: Product Quality Aspects and Storage Stability

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Study Design
2.3. Preparation of Formulations
2.4. Freeze-Drying Process
2.4.1. Conventional Freeze-Drying (CFD)
2.4.2. Microwave-Assisted Freeze-Drying (MFD)
2.5. Karl Fischer Titration
2.6. Brunauer-Emmet-Teller Krypton Gas Adsorption
2.7. X-Ray Powder Diffraction
2.8. Reconstitution of Lyophilizates
2.9. High-Performance Size Exclusion Chromatography (HP-SEC)
2.10. High-Performance Cation Exchange Chromatography (HP-CEX)
2.11. Light Obscuration
2.12. Flow-Imaging Microscopy
3. Results
3.1. Solid State
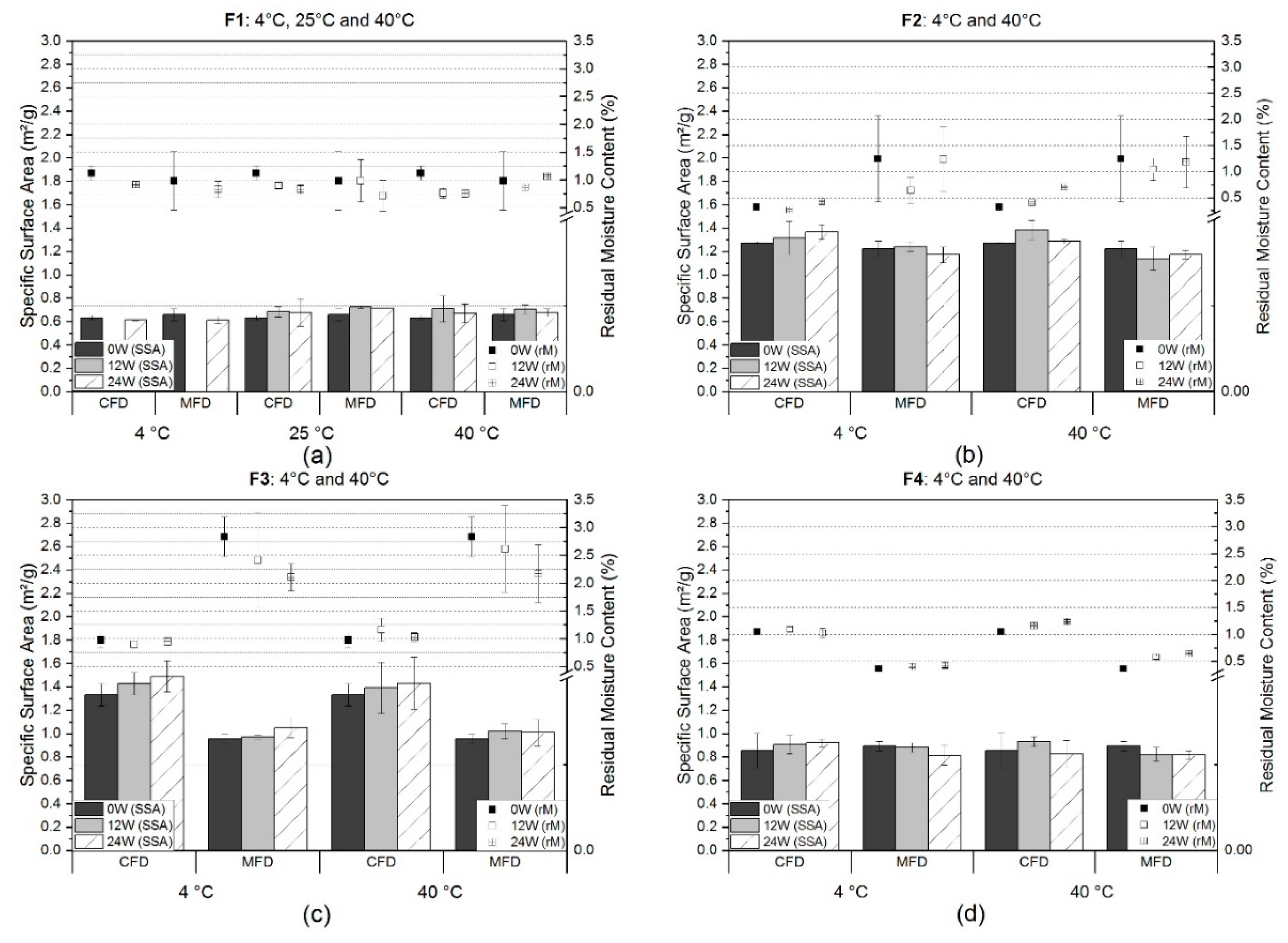
3.1.1. Residual Moisture Content and Specific Surface Area
3.1.2. X-Ray Powder Diffraction (XRD)
3.2. Protein-Related Quality Attributes
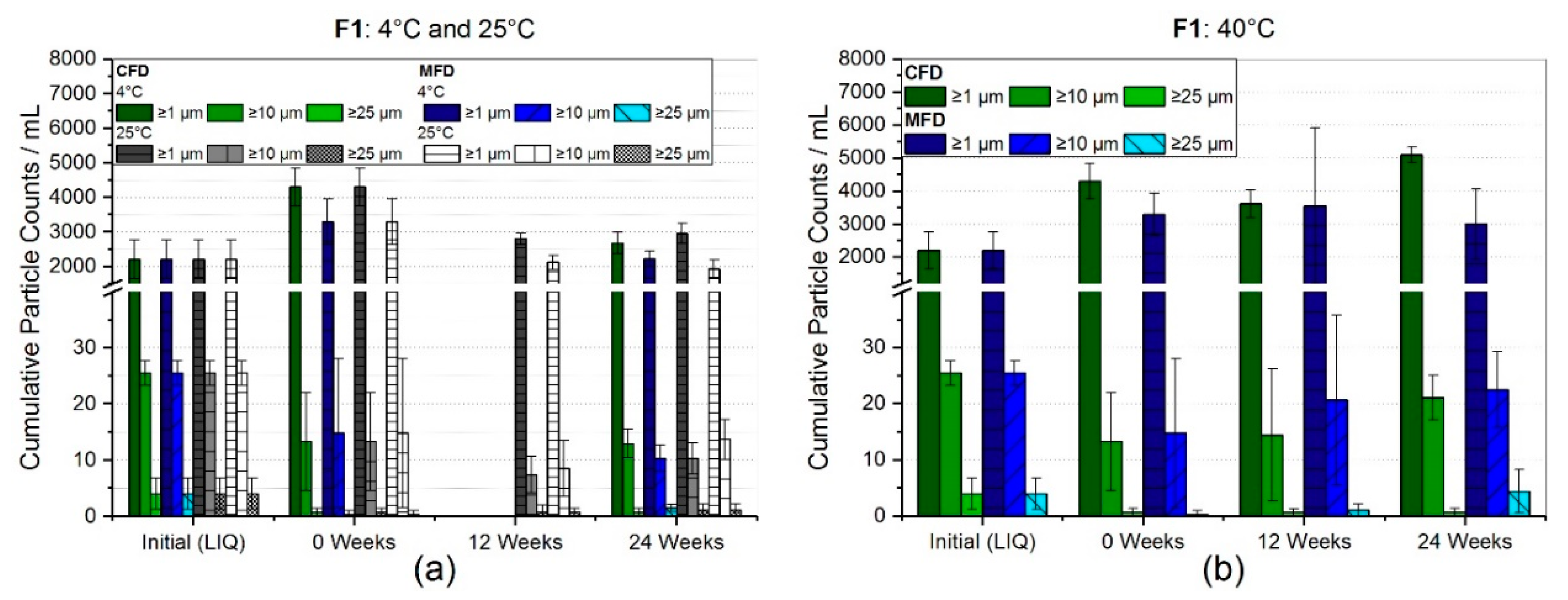
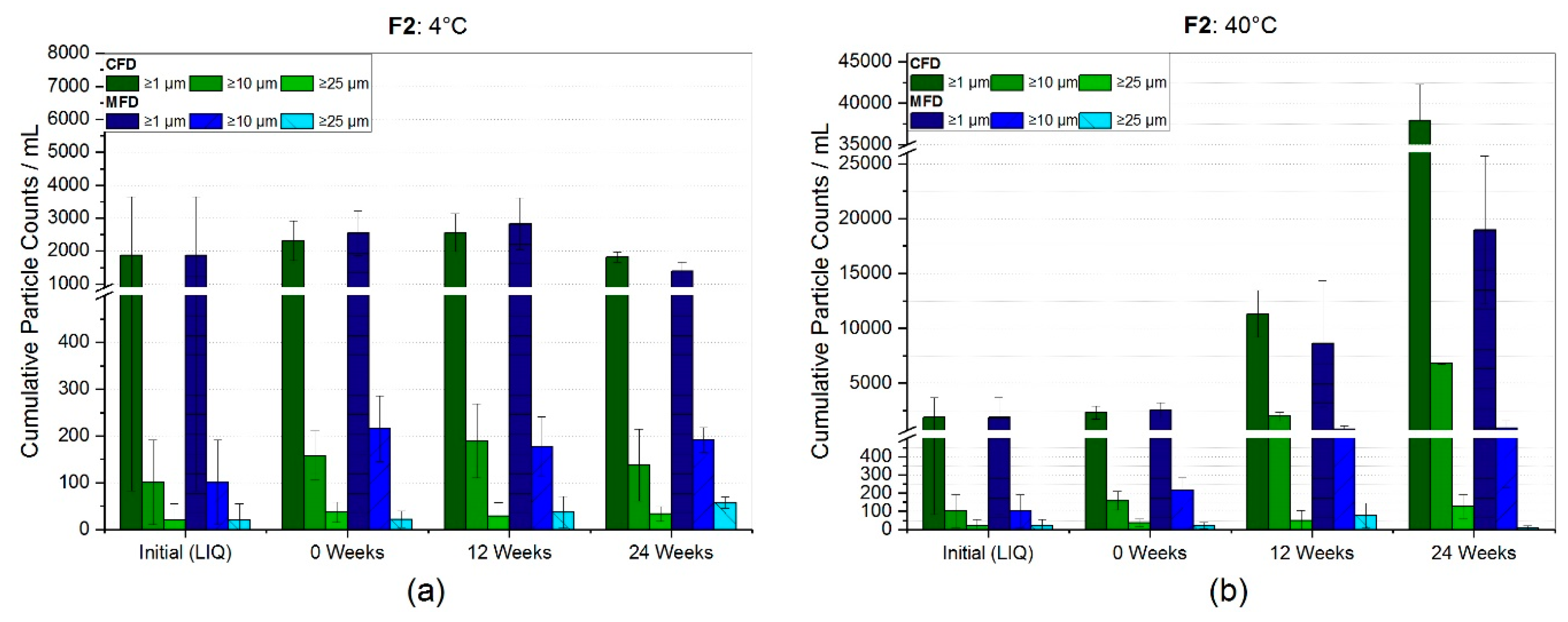
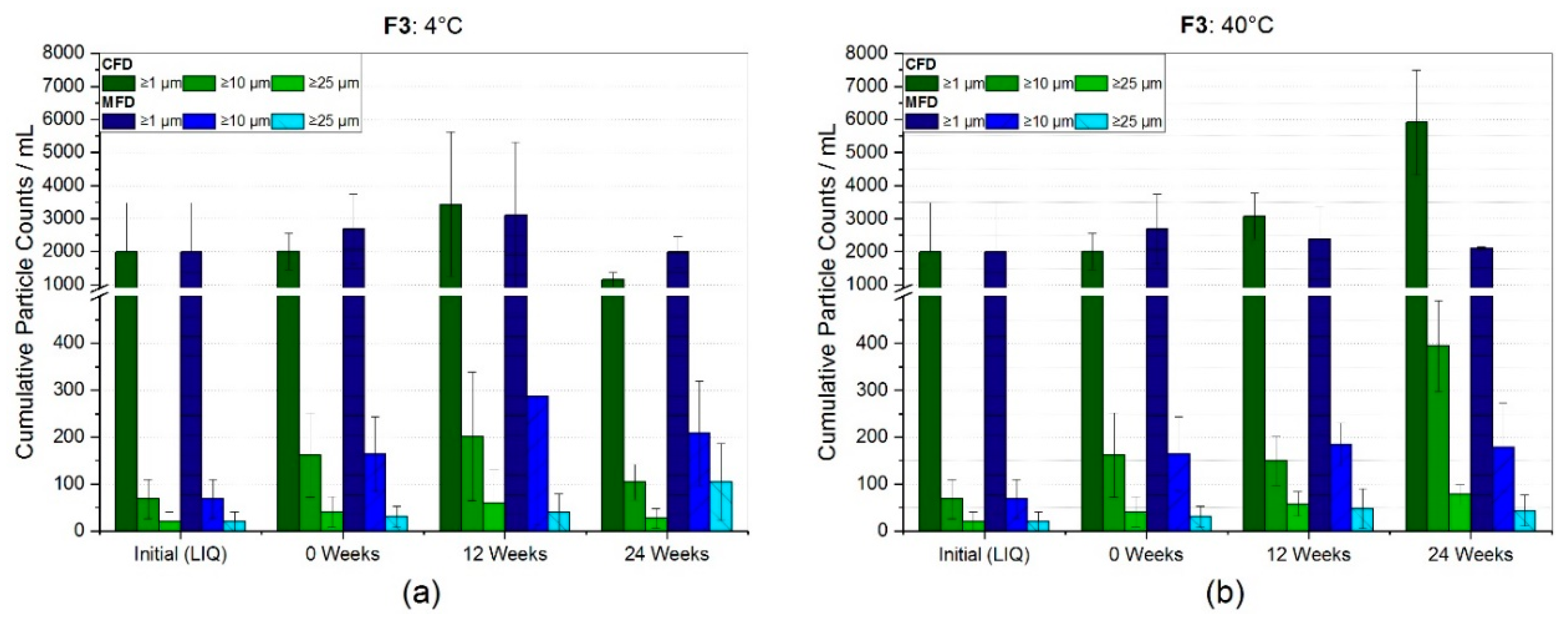
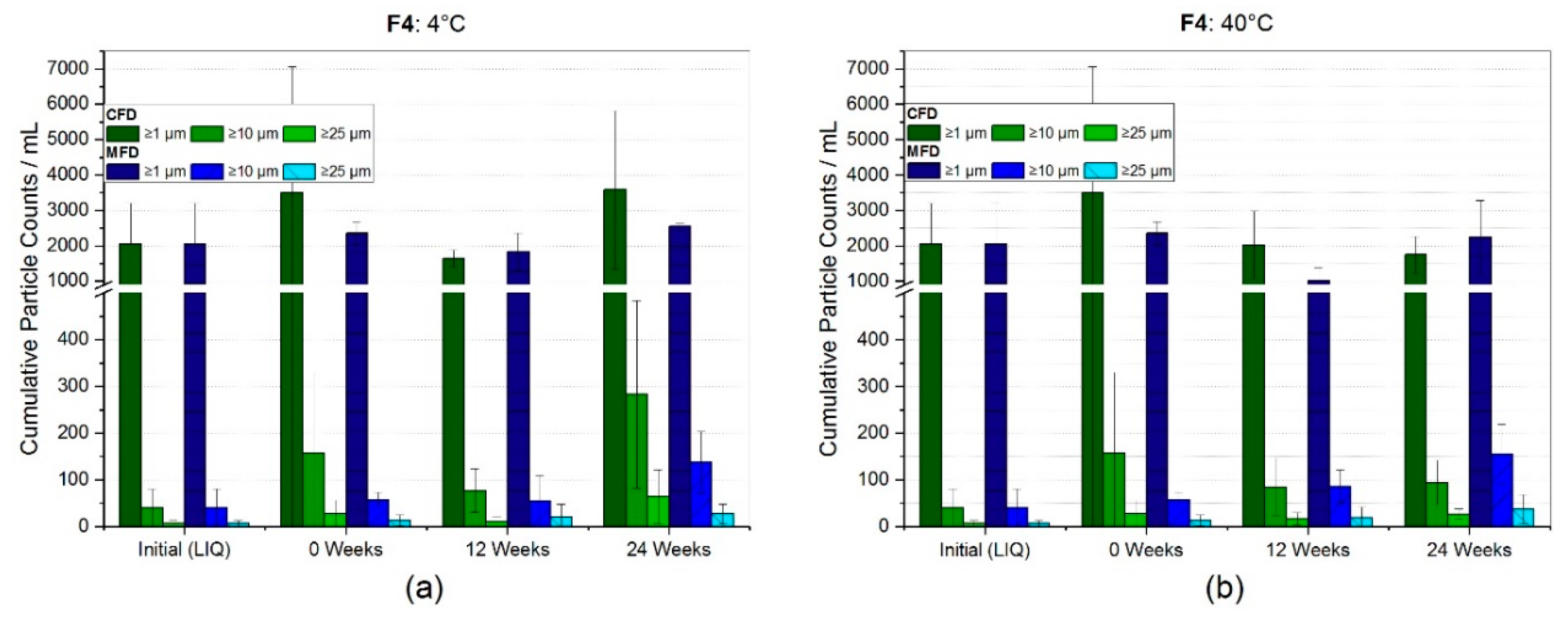
3.2.1. Reconstitution and Subvisible Particles (SvP)
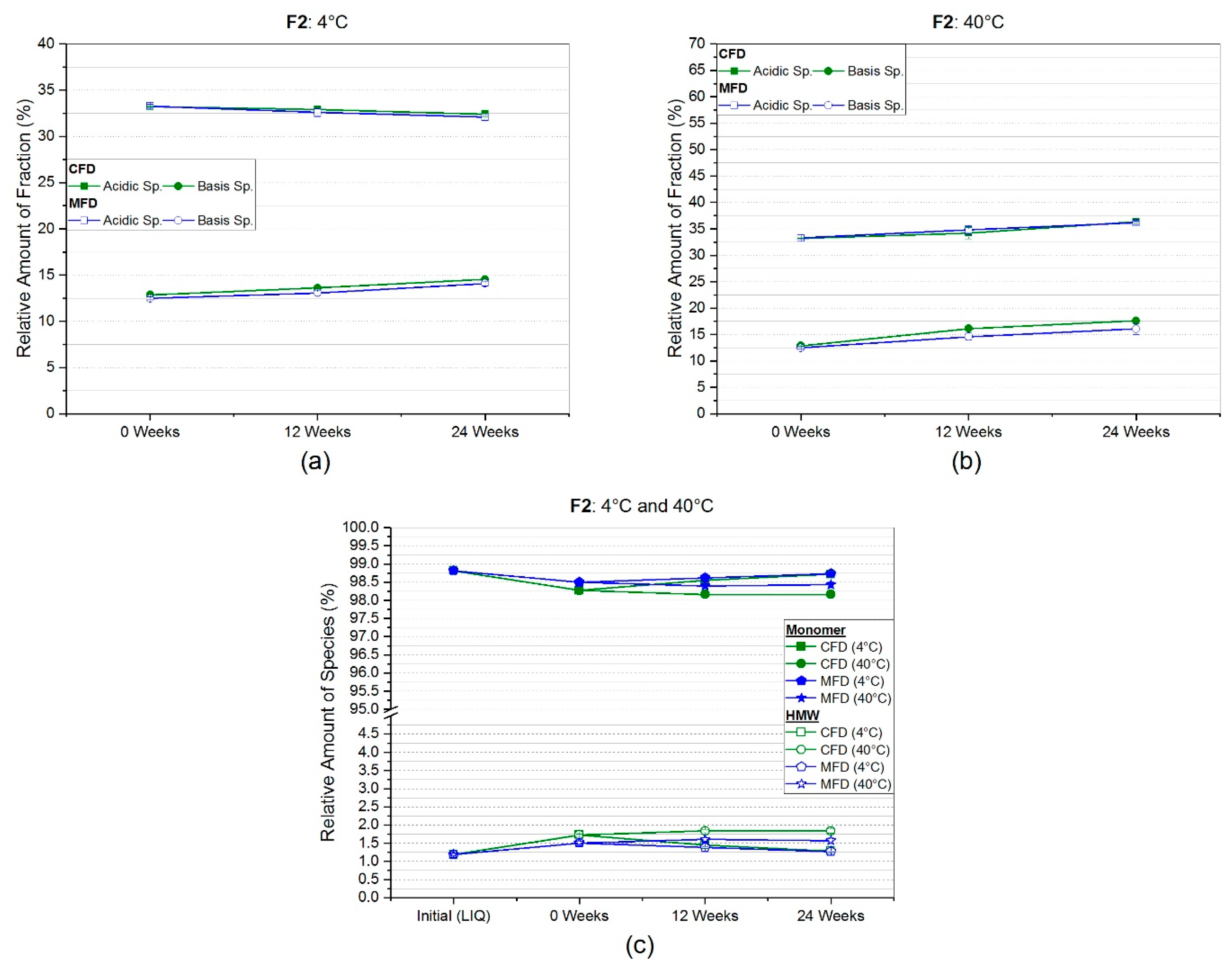
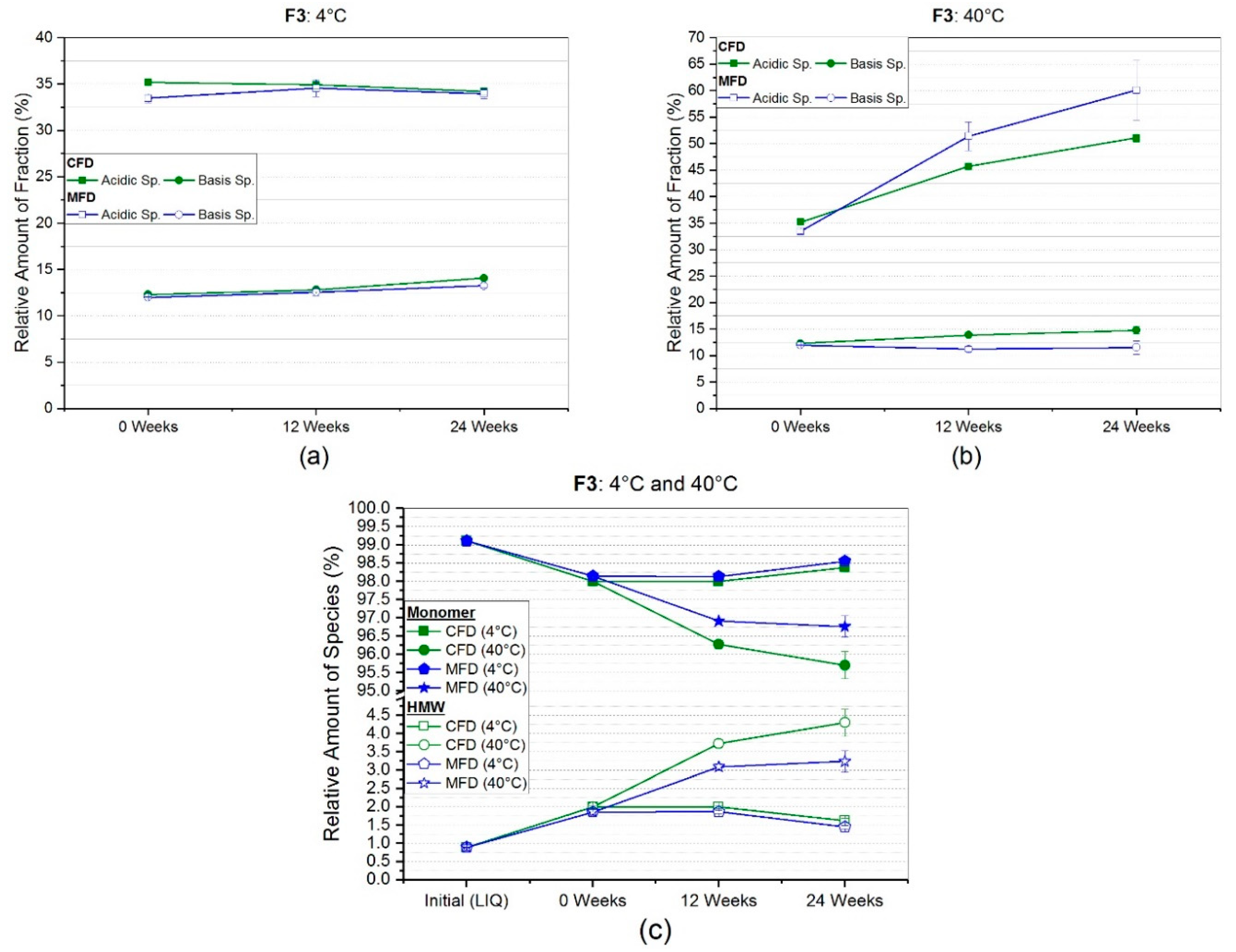
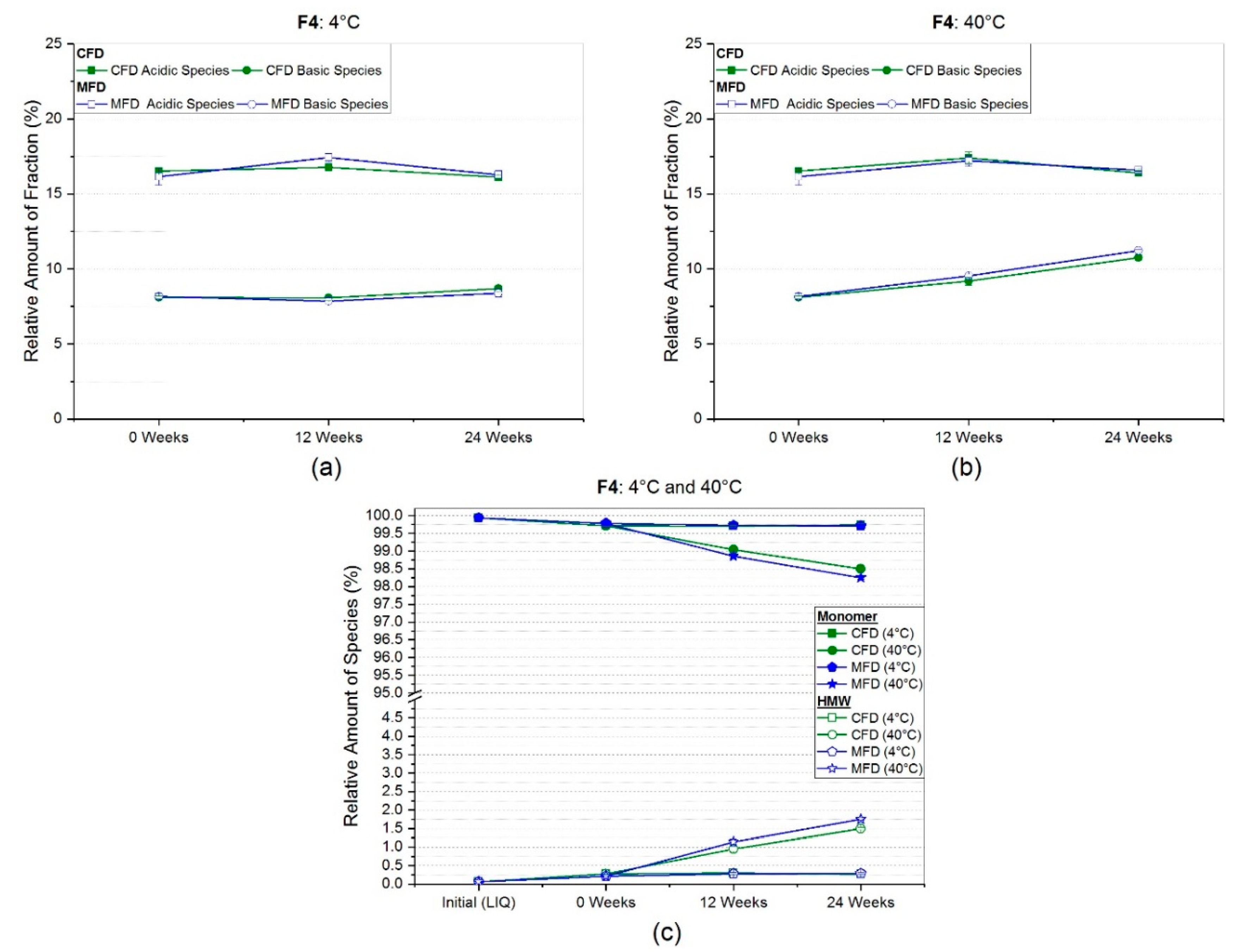
3.2.2. Weak Cation Exchange Chromatography (CEX)
3.2.3. Size Exclusion Chromatography (SEC)
4. Discussion
4.1. Stability with Regard to Solid State Properties
4.2. Stability with Regard to Protein-Related Properties
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carpenter, J.F.; Pikal, M.J.; Chang, B.S.; Randolph, T.W. Rational Design of Stable Lyophilized Protein Formulations: Some Practical Advice. Pharm. Res. 1997, 14, 969. [Google Scholar] [CrossRef] [PubMed]
- Franks, F. Freeze-drying of bioproducts: Putting principles into practice. Eur. J. Pharm. Biopharm. 1998, 45, 221–229. [Google Scholar] [CrossRef]
- Gervasi, V.; Dall Agnol, R.; Cullen, S.; McCoy, T.; Vucen, S.; Crean, A. Parenteral protein formulations: An overview of approved products within the European Union. Eur. J. Pharm. Biopharm. 2018, 131, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Prestrelski, S.J.; Kenney, W.C.; Carpenter, J.F. Factors affecting short-term and long term stabilities of proteins. Adv. Drug Deliv. Rev. 1993, 10, 1–28. [Google Scholar] [CrossRef]
- Franks, F. Long-term stabilization of biologicals. Biotechnology 1994, 12, 253–256. [Google Scholar] [CrossRef]
- Manning, M.C.; Patel, K.; Borchardt, R.T. Stability of Protein Pharmaceuticals. Pharm. Res. 1989, 6, 903–918. [Google Scholar] [CrossRef]
- Manning, M.C.; Chou, D.K.; Murphy, B.M.; Payne, R.W.; Katayama, D.S. Stability of protein pharmaceuticals: An update. Pharm. Res. 2010, 27, 544–575. [Google Scholar] [CrossRef]
- Tang, X.; Pikal, M.J. Design of Freeze-Drying Processes for Pharmaceuticals: Practical Advice. Pharm. Res. 2004, 21, 191–200. [Google Scholar] [CrossRef]
- Patel, S.M.; Pikal, M.J. Emerging freeze-drying process development and scale-up issues. AAPS Pharmscitech 2011, 12, 372–378. [Google Scholar] [CrossRef] [Green Version]
- Bjelošević, M.; Seljak, K.B.; Trstenjak, U.; Logar, M.; Brus, B.; Ahlin Grabnar, P. Aggressive conditions during primary drying as a contemporary approach to optimise freeze-drying cycles of biopharmaceuticals. Eur. J. Pharm. Sci. 2018, 122, 292–302. [Google Scholar] [CrossRef]
- Depaz, R.A.; Pansare, S.; Patel, S.M. Freeze-Drying Above the Glass Transition Temperature in Amorphous Protein Formulations While Maintaining Product Quality and Improving Process Efficiency. J. Pharm. Sci. 2015, 105, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Gitter, J.H.; Geidobler, R.; Presser, I.; Winter, G. A Comparison of Controlled Ice Nucleation Techniques for Freeze-Drying of a Therapeutic Antibody. J. Pharm. Sci. 2018, 107, 2748–2754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jiang, H.; Lim, R.X. Recent developments in microwave-assisted drying of vegetables, fruits, and aquatic products-drying kinetics and quality considerations. Dry. Technol. 2010, 28, 1307–1316. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, M.; Mujumdar, A.S. Physico-chemical changes during different stages of MFD/FD banana chips. J. Food Eng. 2010, 101, 140–145. [Google Scholar] [CrossRef]
- Meredith, R. Engineers’ Handbook of Industrial Microwave Heating; Reprint wi; The Institution of Engineering and Technology: London, UK, 2007; ISBN 978-0-85296-916-8. [Google Scholar]
- Metaxas, A.C. Microwave heating. Power Eng. J. 1991, 5, 237–247. [Google Scholar] [CrossRef]
- Datta, A.K.; Davidson, P.M. Microwave and Radio Frequency Processing. J. Food Sci. 2000, 65, 32–41. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Ramanathan, S.; Basak, T. Microwave food processing—A review. Food Res. Int. 2013, 52, 243–261. [Google Scholar] [CrossRef]
- Ambros, S.; Mayer, R.; Schumann, B.; Kulozik, U. Microwave-freeze drying of lactic acid bacteria: Influence of process parameters on drying behavior and viability. Innov. Food Sci. Emerg. Technol. 2018, 48, 90–98. [Google Scholar] [CrossRef]
- Gitter, J.H.; Geidobler, R.; Presser, I.; Winter, G. Significant Drying Time Reduction Using Microwave-Assisted Freeze-Drying for a Monoclonal Antibody. J. Pharm. Sci. 2018, 107, 2538–2543. [Google Scholar] [CrossRef]
- Duan, X.; Zhang, M.; Mujumdar, A.S.; Wang, S. Microwave freeze drying of sea cucumber (Stichopus japonicus). J. Food Eng. 2010, 96, 491–497. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, M.; Liu, Y.; Mujumdar, A.S.; Liu, H. The energy consumption and color analysis of freeze/microwave freeze banana chips. Food Bioprod. Process. Trans. Inst. Chem. Eng. Part C 2013, 91, 464–472. [Google Scholar] [CrossRef]
- Duan, X.; Zhang, M.; Mujumdar, A.S. Studies on the Microwave Freeze Drying Technique and Sterilization Characteristics of Cabbage. Dry. Technol. 2007, 25, 1725–1731. [Google Scholar] [CrossRef]
- Evans, R. Applications of Microwave Vacuum Drying and Lyospheres to Freeze-Drying of Vaccines and Biologics. In Proceedings of the CPPR: Freeze-Drying of Pharmaceuticals and Biologicals Conference, Garmisch-Partenkrichen, Germany, 23–26 September 2014. [Google Scholar]
- Durance, T.D.; Fu, J. Apparatus and Method for Dehydrating Biological Materials. World Intellectual Property Organization Patent Application No. WO2010124375A1, 4 November 2010. [Google Scholar]
- Bhambhani, A.; Evans, R.K.; Sinacola, J.; Jones, M. Method of Obtaining Thermostable Dried Vaccine Formulations. World Intellectual Property Organization Patent Application No. WO 2015/057541 A1, 23 April 2015. [Google Scholar]
- Bhambhani, A.; Evans, R.K.; Sinacola, J.; Lizzano, R. Method of Microwave Vacuum Drying Spherical-shaped Pellets of Biological Materials. World Intellectual Property Organization Patent Application No. WO2015057540A1, 23 April 2015. [Google Scholar]
- Püschner GmbH & Co. KG. Good Manufacturing Practise (GMP) for Industrial Microwave Plants. Available online: http://www.pueschner.com/en/microwave-technology/gmp-microwaves (accessed on 10 April 2018).
- Sundaramurthi, P.; Suryanarayanan, R. Trehalose crystallization during freeze-drying: Implications on lyoprotection. J. Phys. Chem. Lett. 2010, 1, 510–514. [Google Scholar] [CrossRef]
- Sundaramurthi, P.; Suryanarayanan, R. Influence of crystallizing and non-crystallizing cosolutes on trehalose crystallization during freeze-drying. Pharm. Res. 2010, 27, 2384–2393. [Google Scholar] [CrossRef]
- Liu, J. Physical characterization of pharmaceutical formulations in frozen and freeze-dried solid states: Techniques and applications in freeze-drying development. Pharm. Dev. Technol. 2006, 11, 3–28. [Google Scholar] [CrossRef]
- Searles, J.A.; Carpenter, J.F.; Randolph, T.W. The ice nucleation temperature determines the primary drying rate of lyophilization for samples frozen on a temperature-controlled shelf. J. Pharm. Sci. 2001, 90, 860–871. [Google Scholar] [CrossRef]
- Vadivambal, R.; Jayas, D.S. Non-uniform temperature distribution during microwave heating of food materials—A review. Food Bioprocess Technol. 2010, 3, 161–171. [Google Scholar] [CrossRef]
- Burfoot, D.; Griffin, W.J.; James, S.J. Microwave pasteurisation of prepared meals. J. Food Eng. 1988, 8, 145–156. [Google Scholar] [CrossRef]
- Vilayannur, R.S.; Puri, V.M.; Anantheswaran, R.C. Size and shape effect on nonuniformity of temperature and moisture distributions in microwave heated food materials: Part II experimental validation. J. Food Process Eng. 1998, 21, 235–248. [Google Scholar] [CrossRef]
- Zhang, Z.; Su, T.; Zhang, S. Shape Effect on the Temperature Field during Microwave Heating Process. J. Food Qual. 2018, 2018, 1–24. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Swain, M.V.; James, S.J.; Swain, M.J. Development of methodology for assessing the heating performance of domestic microwave ovens. Int. J. Food Sci. Technol. 2002, 37, 879–892. [Google Scholar] [CrossRef]
- Rattanadecho, P. Theoretical and experimental investigation of microwave thawing of frozen layer using a microwave oven (effects of layered configurations and layer thickness). Int. J. Heat Mass Transf. 2004, 47, 937–945. [Google Scholar] [CrossRef]
- Lobo, S.; Datta, A.K. Characterization of Spatial Non-Uniformity in Microwave Reheating of High Loss Foods. J. Microw. Power Electromagn. Energy 1998, 33, 158–166. [Google Scholar] [CrossRef]
- Pikal, M.J.; Shah, S. Moisture transfer from stopper to product and resulting stability implications. Dev. Biol. Stand. 1992, 74, 165–177. [Google Scholar] [PubMed]
- Meng, C.; Da-ming, F.; Lue-lue, H.; Yi-shu, G.; Jian-lian, H.; Jian-xin, Z.; Hao, Z. A new approach to microwave food research: Analyzing the electromagnetic response of basic amino acids. Innov. Food Sci. Emerg. Technol. 2017, 41, 100–108. [Google Scholar] [CrossRef]
- Aoki, K.; Nagano, K.; Iitaka, Y. The crystal structure of L-arginine phosphate monohydrate. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1971, 27, 11–23. [Google Scholar] [CrossRef]
- Izutsu, K.I.; Fujimaki, Y.; Kuwabara, A.; Aoyagi, N. Effect of counterions on the physical properties of L-arginine in frozen solutions and freeze-dried solids. Int. J. Pharm. 2005, 301, 161–169. [Google Scholar] [CrossRef]
- Stärtzel, P.; Gieseler, H.; Gieseler, M.; Abdul-Fattah, A.M.; Adler, M.; Mahler, H.-C.; Goldbach, P. Freeze Drying of L-Arginine/Sucrose-Based Protein Formulations, Part I: Influence of Formulation and Arginine Counter Ion on the Critical Formulation Temperature, Product Performance and Protein Stability. J. Pharm. Sci. 2015, 104, 2345–2358. [Google Scholar] [CrossRef]
- Pikal, M.J.; Shah, S.; Roy, M.L.; Putman, R. The secondary drying stage of freeze drying: Drying kinetics as a function of temperature and chamber pressure. Int. J. Pharm. 1990, 60, 203–217. [Google Scholar] [CrossRef]
- Bianchi, C.; Schmid, R.; Frick, D.; Kurtz, S. Microwave Assisted Vacuum Drying Processing: Magnetron vs Solid State. Case Study: Apple Drying. In Proceedings of the COMSOL Conference, Lausanne, Switzerland, 22–24 October 2018. [Google Scholar]
- Wang, W. Lyophilization and development of solid protein pharmaceuticals. Int. J. Pharm. 2000, 203, 1–60. [Google Scholar] [CrossRef]
- Du, Y.; Walsh, A.; Ehrick, R.; Xu, W.; May, K.; Liu, H. Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies. MAbs 2012, 4, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Fekete, S.; Beck, A.; Veuthey, J.L.; Guillarme, D. Ion-exchange chromatography for the characterization of biopharmaceuticals. J. Pharm. Biomed. Anal. 2015, 113, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.J.; Kabakoff, B.; Macchi, F.D.; Shen, F.J.; Kwong, M.; Andya, J.D.; Shire, S.J.; Bjork, N.; Totpal, K.; Chen, A.B. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J. Chromatogr. B Biomed. Sci. Appl. 2001, 752, 233–245. [Google Scholar] [CrossRef]
- Stärtzel, P. Arginine as an Excipient for Protein Freeze-Drying: A Mini Review. J. Pharm. Sci. 2018, 107, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Fattah, A.M.; Truong-Le, V.; Yee, L.; Nguyen, L.; Kalonia, D.S.; Cicerone, M.T.; Pikal, M.J. Drying-Induced Variations in Physico-Chemical Properties of Amorphous Pharmaceuticals and Their Impact on Stability (I): Stability of a Monoclonal Antibody. J. Pharm. Sci. 2007, 96, 1983–2008. [Google Scholar] [PubMed]
- Craig, D.Q.M.; Royall, P.G.; Kett, V.L.; Hopton, M.L. The relevance of the amorphous state to pharmaceutical dosage forms: Glassy drugs and freeze dried systems. Int. J. Pharm. 1999, 179, 179–207. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, S.; Aso, Y. Correlations between Molecular Mobility and Chemical Stability during Storage of Amorphous Pharmaceuticals. J. Pharm. Sci. 2007, 96, 960–981. [Google Scholar] [CrossRef]
- Abdul-Fattah, A.M.; Kalonia, D.S.; Pikal, M.J. The Challenge of Drying Method Selection for Protein Pharmaceuticals: Product Quality Implications. J. Pharm. Sci. 2007, 96, 1886–1916. [Google Scholar] [CrossRef]
- Schersch, K.; Betz, O.; Garidel, P.; Muehlau, S.; Bassarab, S.; Winter, G. Systematic Investigation of the Effect of Lyophilizate Collapse on Pharmaceutically Relevant Proteins, Part 2: Stability During Storage at Elevated Temperatures. J. Pharm. Sci. 2012, 101, 2288–2306. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredient | F1 | F2 | F3 | F4 |
|---|---|---|---|---|
| mAb1 [g/L] | 5 | 5 | 5 | / |
| mAb2 [g/L] | / | / | / | 50 |
| Sucrose [% (w/v] | 10 | / | / | 5 |
| Trehalose [% (w/v)] | / | 10 | / | / |
| Arginine phosphate [% (w/v)] | / | / | 10 | / |
| Polysorbate 80 [% (w/v)] | 0.02 | 0.02 | 0.02 | / |
| Freeze-Drying Process | Setpoint | Freezing | Primary Drying | Secondary Drying | |||
|---|---|---|---|---|---|---|---|
| CFD cycle F1 | TShelf [°C] | 20 | −5 | −60 | −20 | 0 | 20 |
| Ramp [K/min] | − | 1 | 1 | 0.2 | 0.05 | 0.2 | |
| Hold time [min] | 5 | 60 | 2580 a | 2700 (1120) b | − | 360 | |
| pChamber [µbar] | − | − | − | 100 | 50 | 50 | |
| CFD cycle F2/3 | TShelf [°C] | 20 | −5 | −60 | −25 | 0 | 20 |
| Ramp [K/min] | − | 1 | 1 | 0.2 | 0.05 | 0.2 | |
| Hold time [min] | 5 | 60 | 3610 a | 5760 (1400) b | - | 360 | |
| pChamber [mbar] | − | - | − | 100 | 50 | 50 | |
| CFD cycle F4 | TShelf [°C] | 20 | 5 | −50 | −20 | 5 | 35 |
| Ramp [K/min] | − | 1 | 1 | 1 | 0.15 | 0.3 | |
| Hold time [min] | 5 | 60 | 1006 a | 1868 (1058) b | − | 420 | |
| pChamber [mbar] | − | − | − | 100 | 100 | 100 | |
| Formulation | Reconstitution Time (s) |
|---|---|
| F1 | ≤30 |
| F2 | ≤30 |
| F3 | ≤50 |
| F4 | ≤120 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gitter, J.H.; Geidobler, R.; Presser, I.; Winter, G. Microwave-Assisted Freeze-Drying of Monoclonal Antibodies: Product Quality Aspects and Storage Stability. Pharmaceutics 2019, 11, 674. https://doi.org/10.3390/pharmaceutics11120674
Gitter JH, Geidobler R, Presser I, Winter G. Microwave-Assisted Freeze-Drying of Monoclonal Antibodies: Product Quality Aspects and Storage Stability. Pharmaceutics. 2019; 11(12):674. https://doi.org/10.3390/pharmaceutics11120674
Chicago/Turabian StyleGitter, Julian Hendryk, Raimund Geidobler, Ingo Presser, and Gerhard Winter. 2019. "Microwave-Assisted Freeze-Drying of Monoclonal Antibodies: Product Quality Aspects and Storage Stability" Pharmaceutics 11, no. 12: 674. https://doi.org/10.3390/pharmaceutics11120674