Controlling and Predicting the Dissolution Kinetics of Thermally Oxidised Mesoporous Silicon Particles: Towards Improved Drug Delivery

Abstract
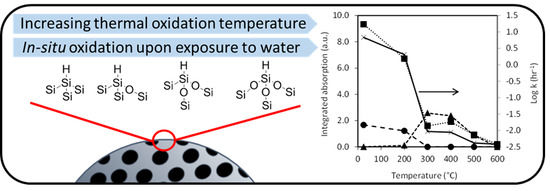
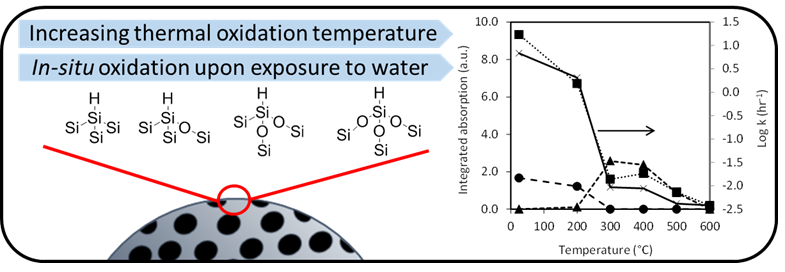
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. In Vitro pSi Dissolution
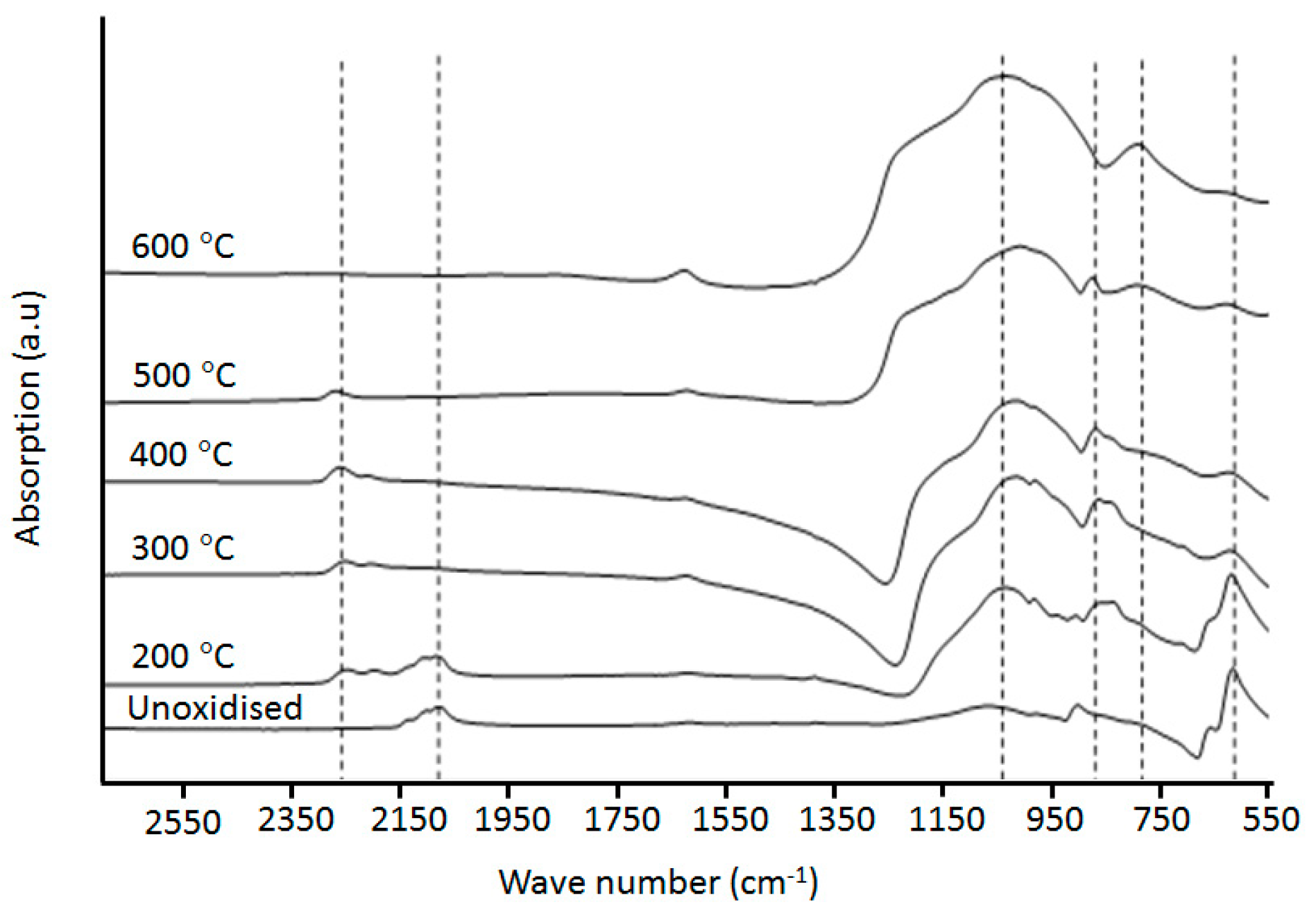
2.3. Fourier Transform Infrared (FTIR) Spectroscopy
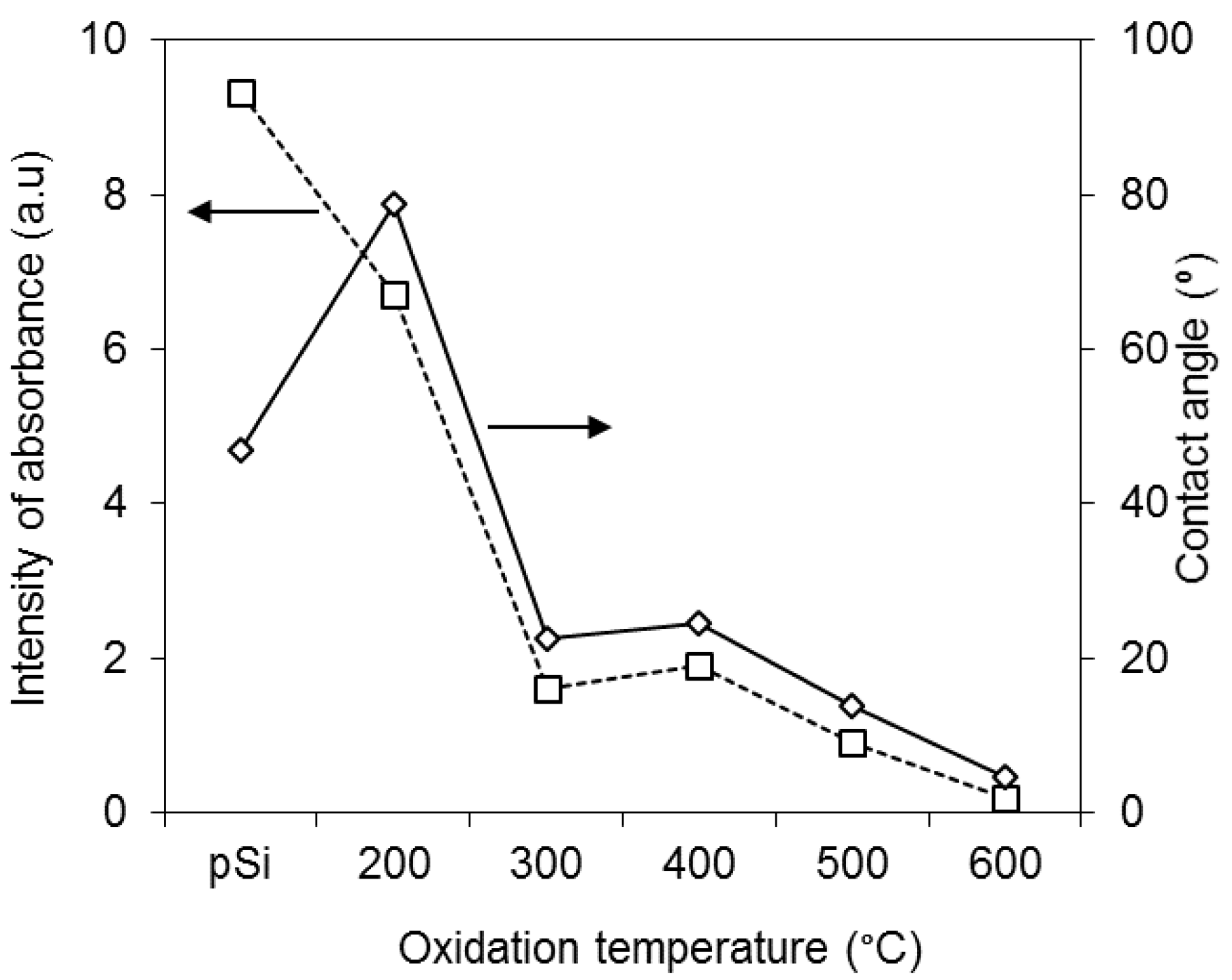
2.4. Contact Angle Measurements
3. Results
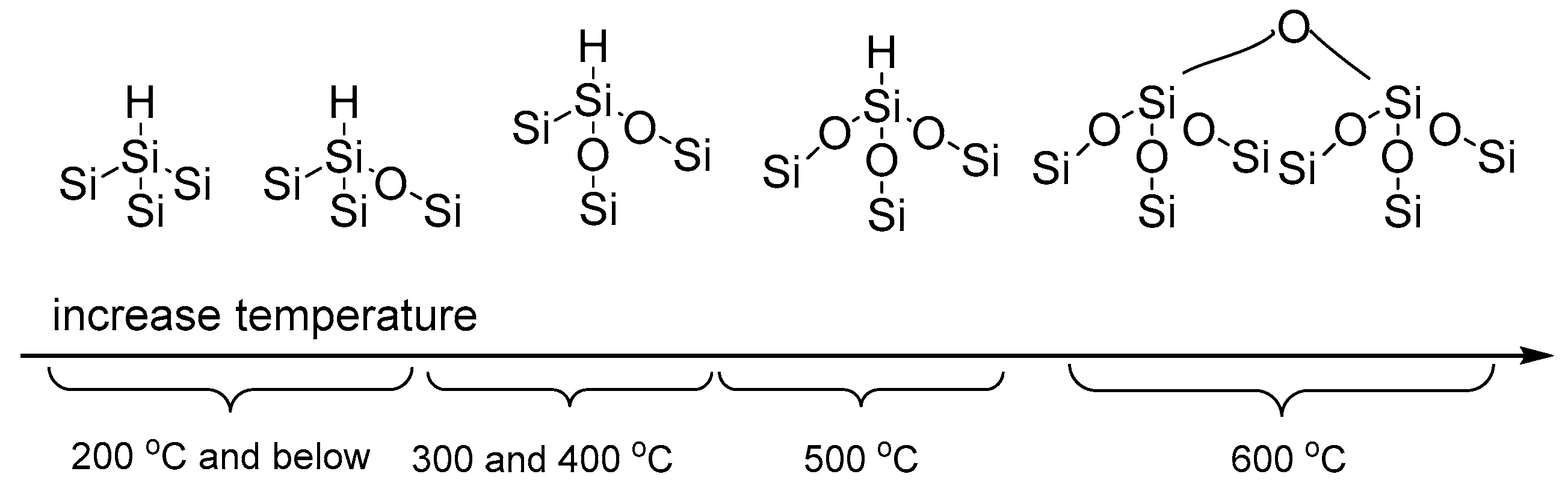
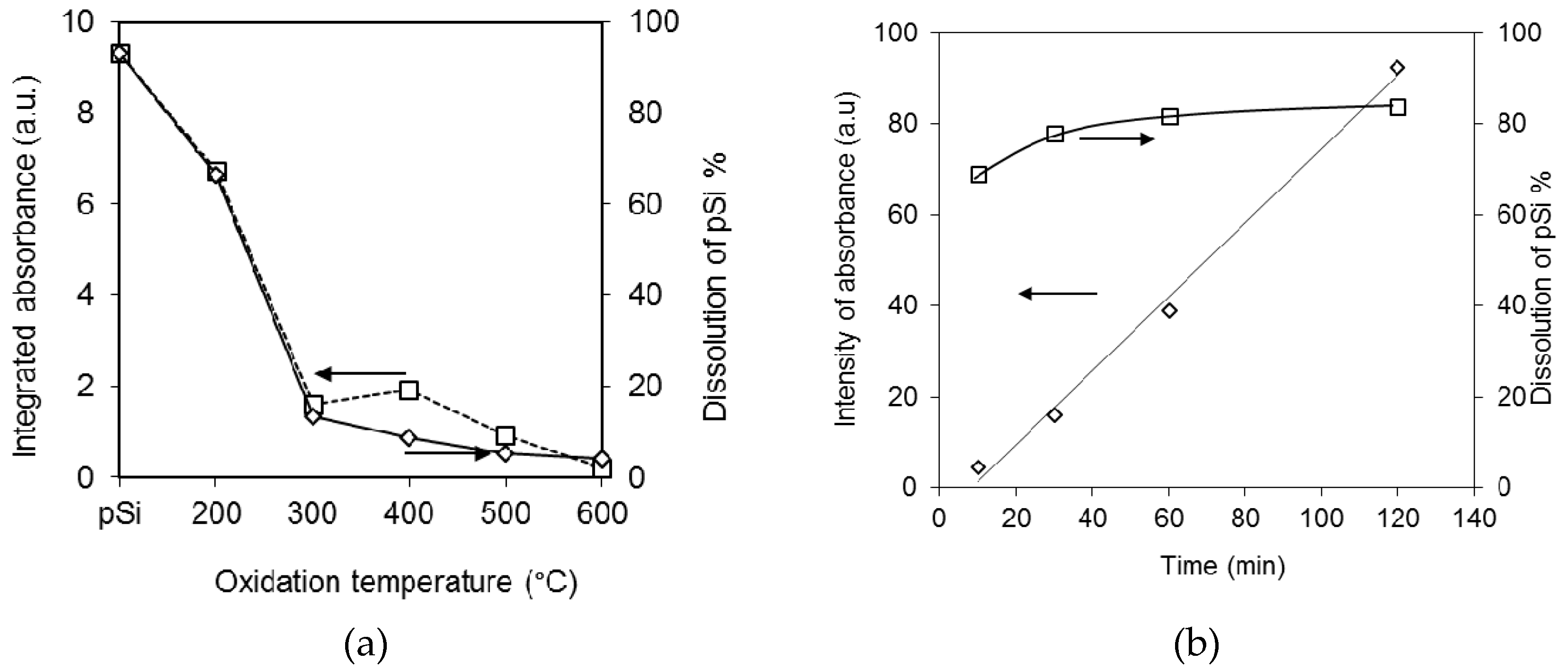
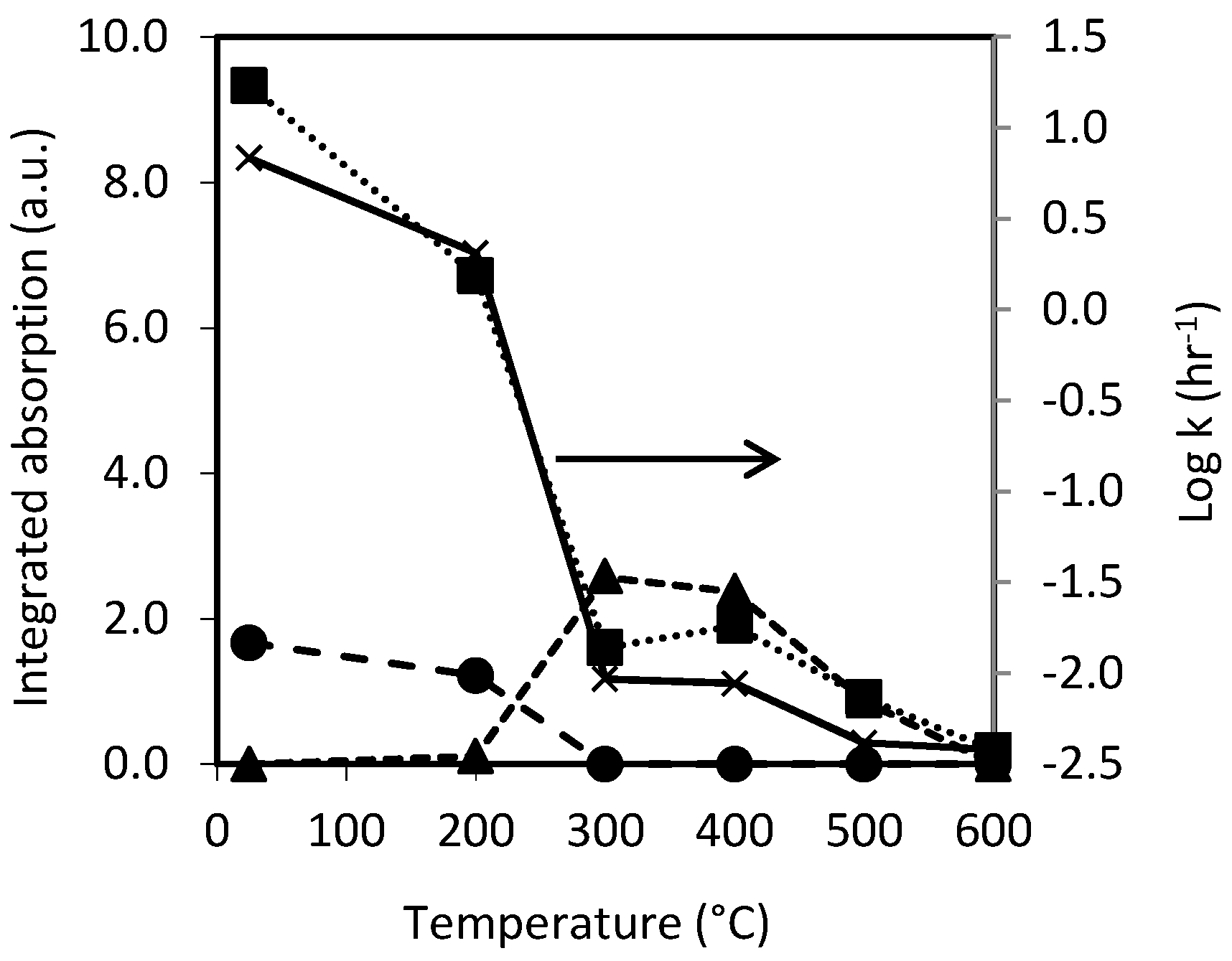
3.1. Preparation and Characterisation of Thermally Oxidised pSi Microparticles
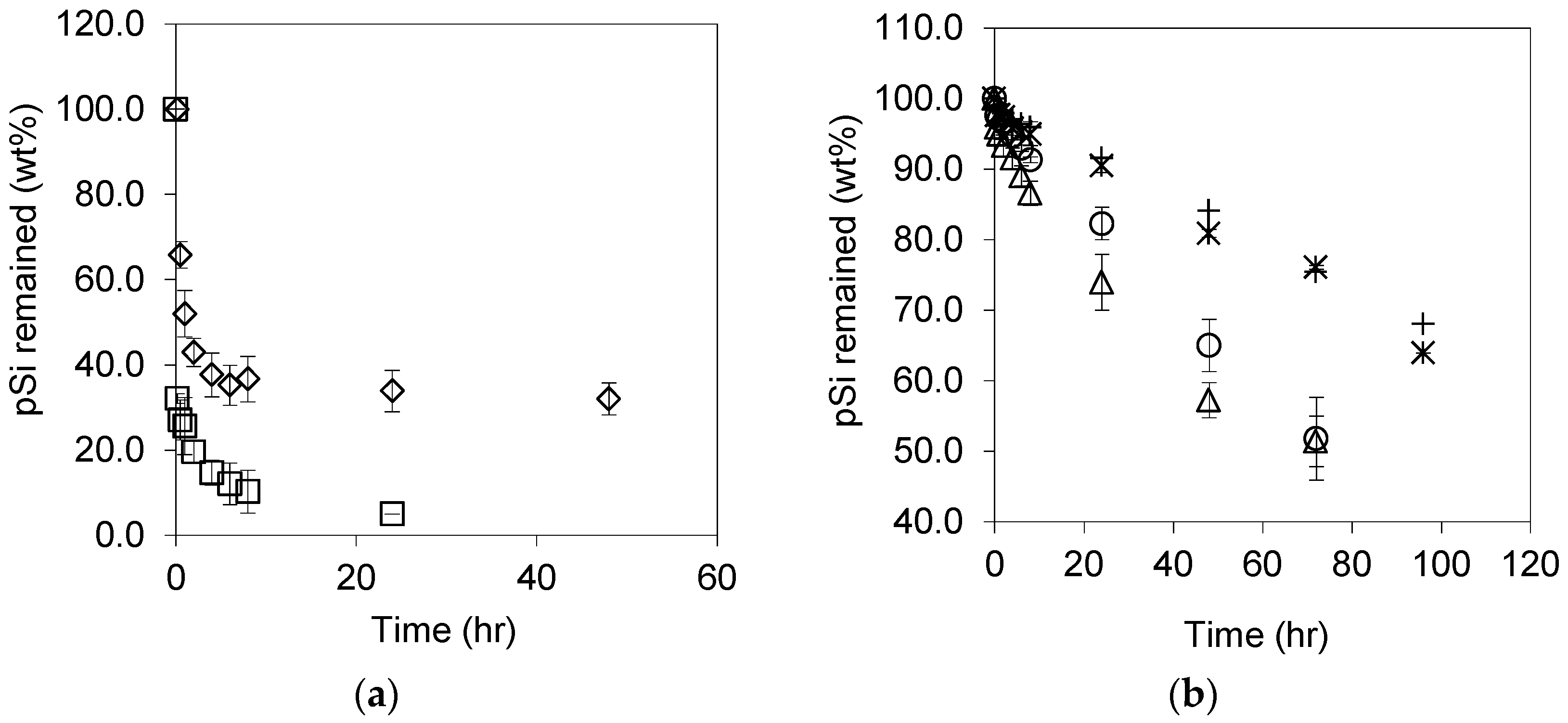
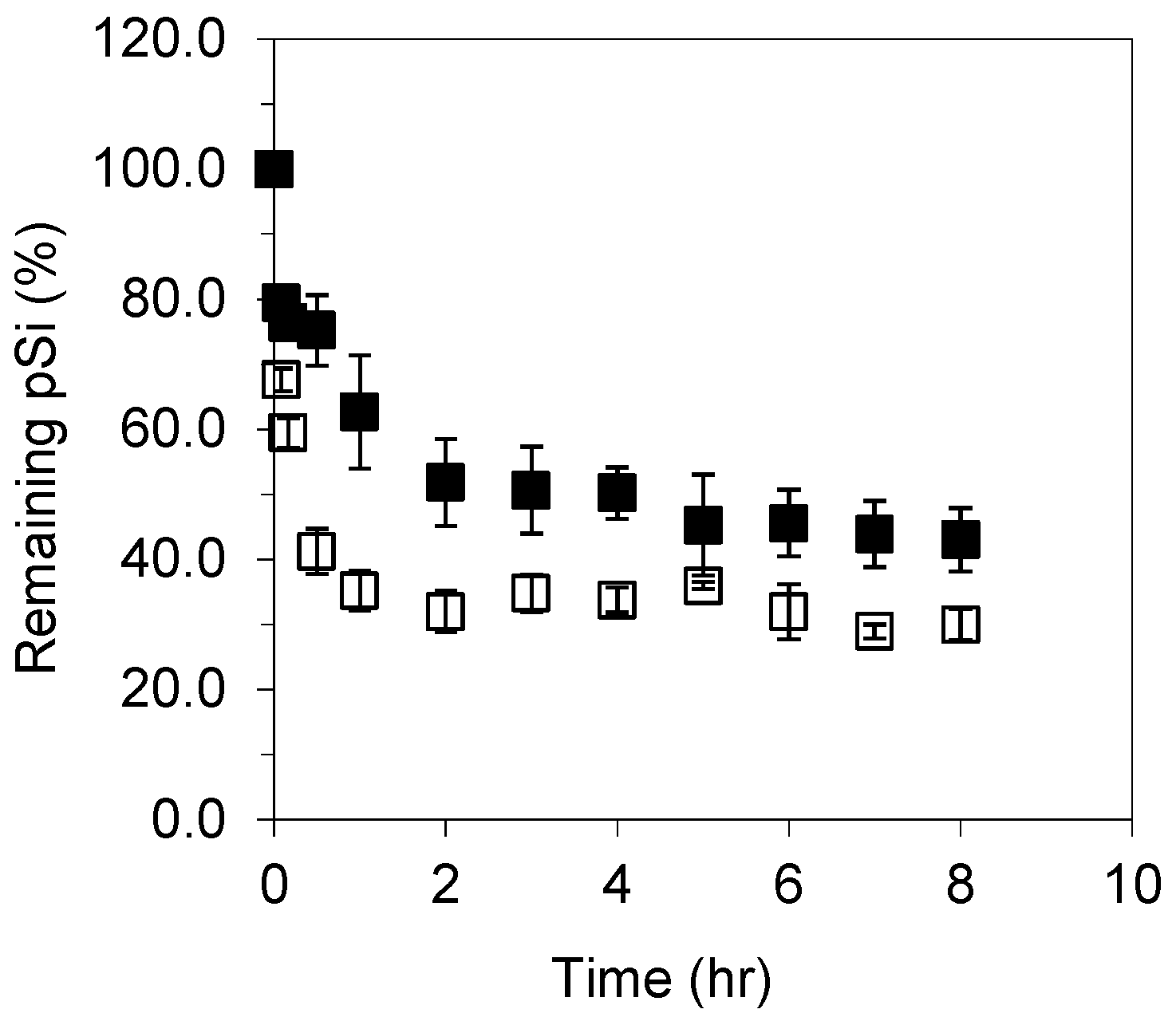
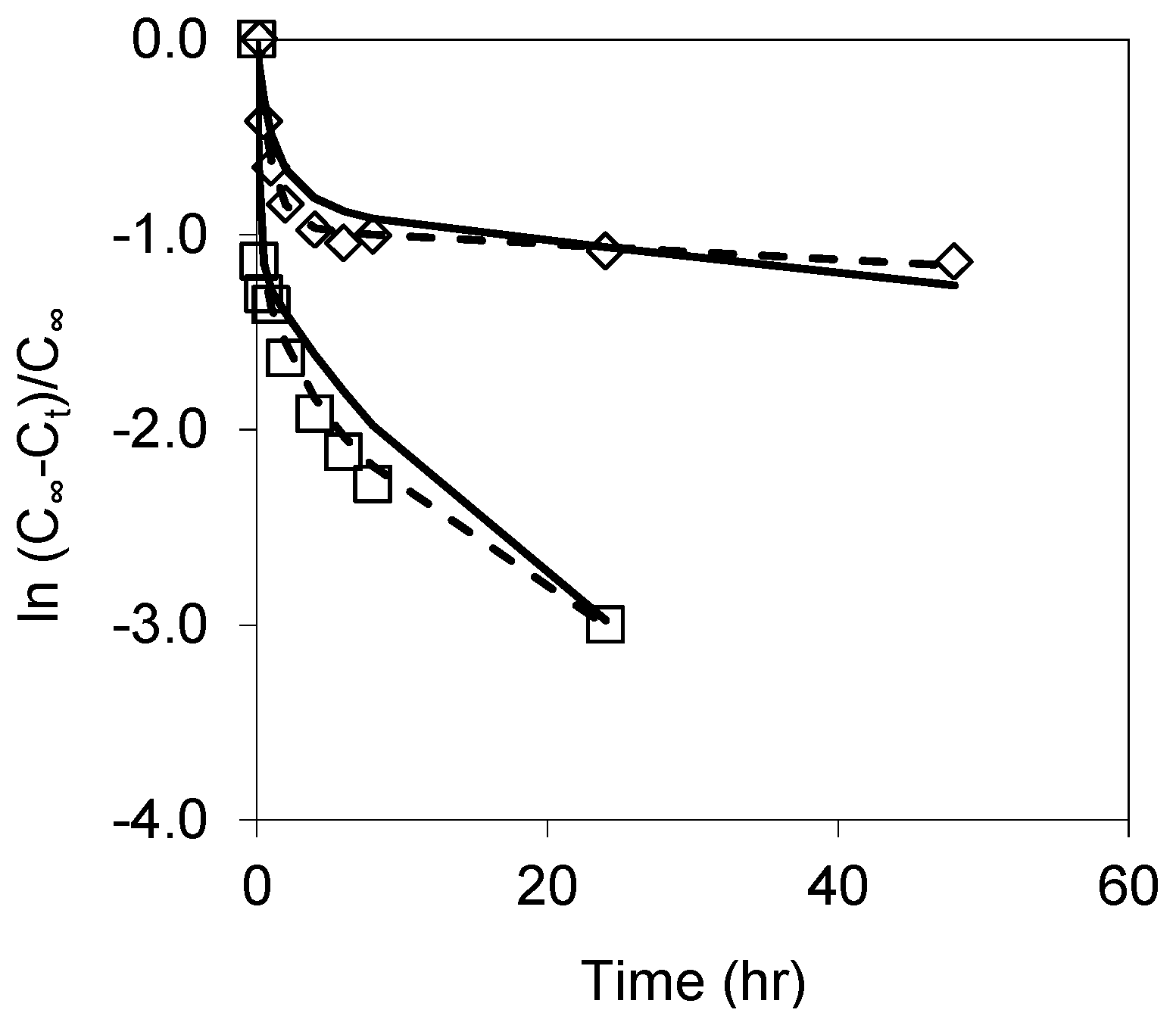
3.2. Dissolution of Unoxidized and Thermally Oxidised pSi
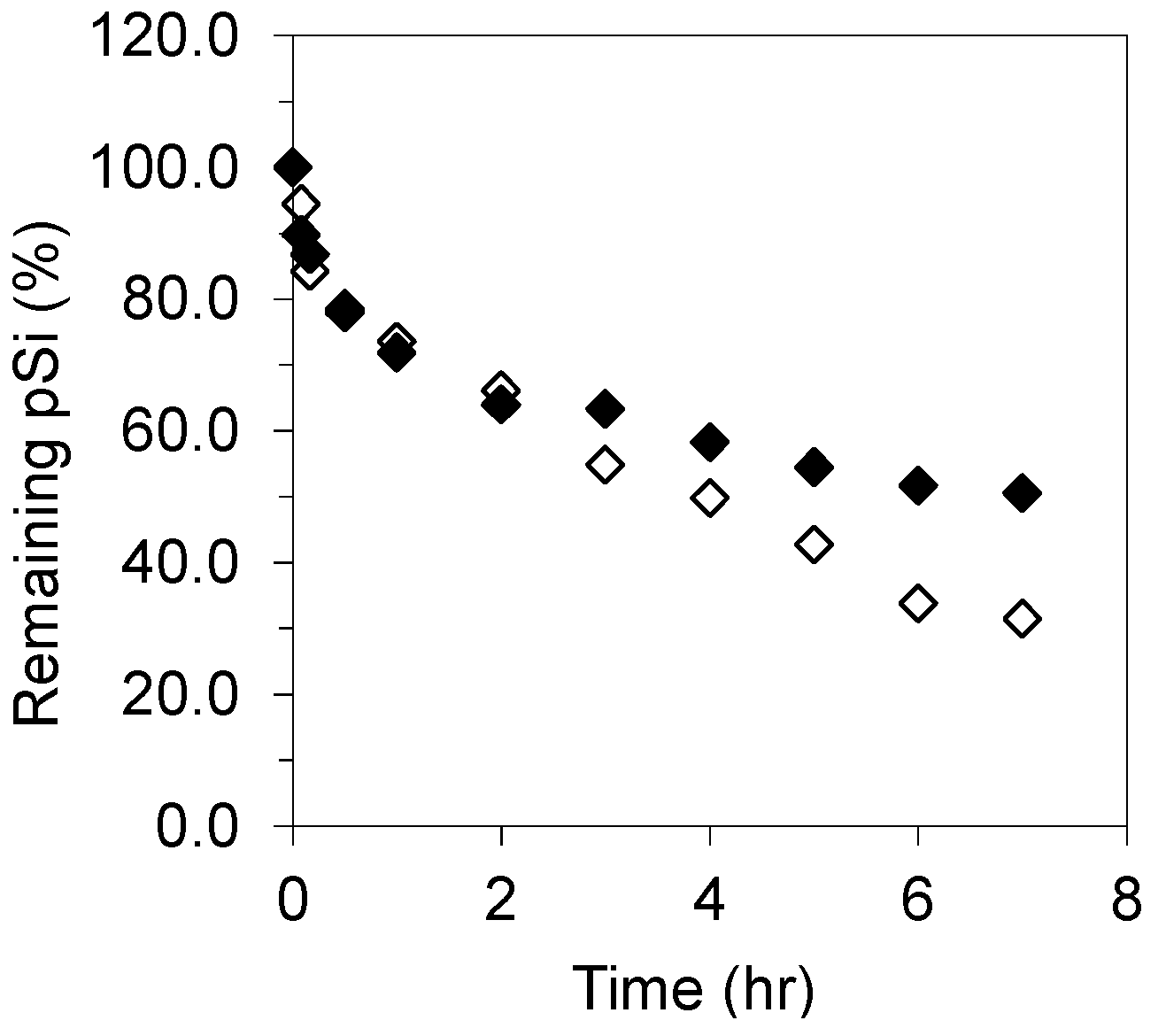
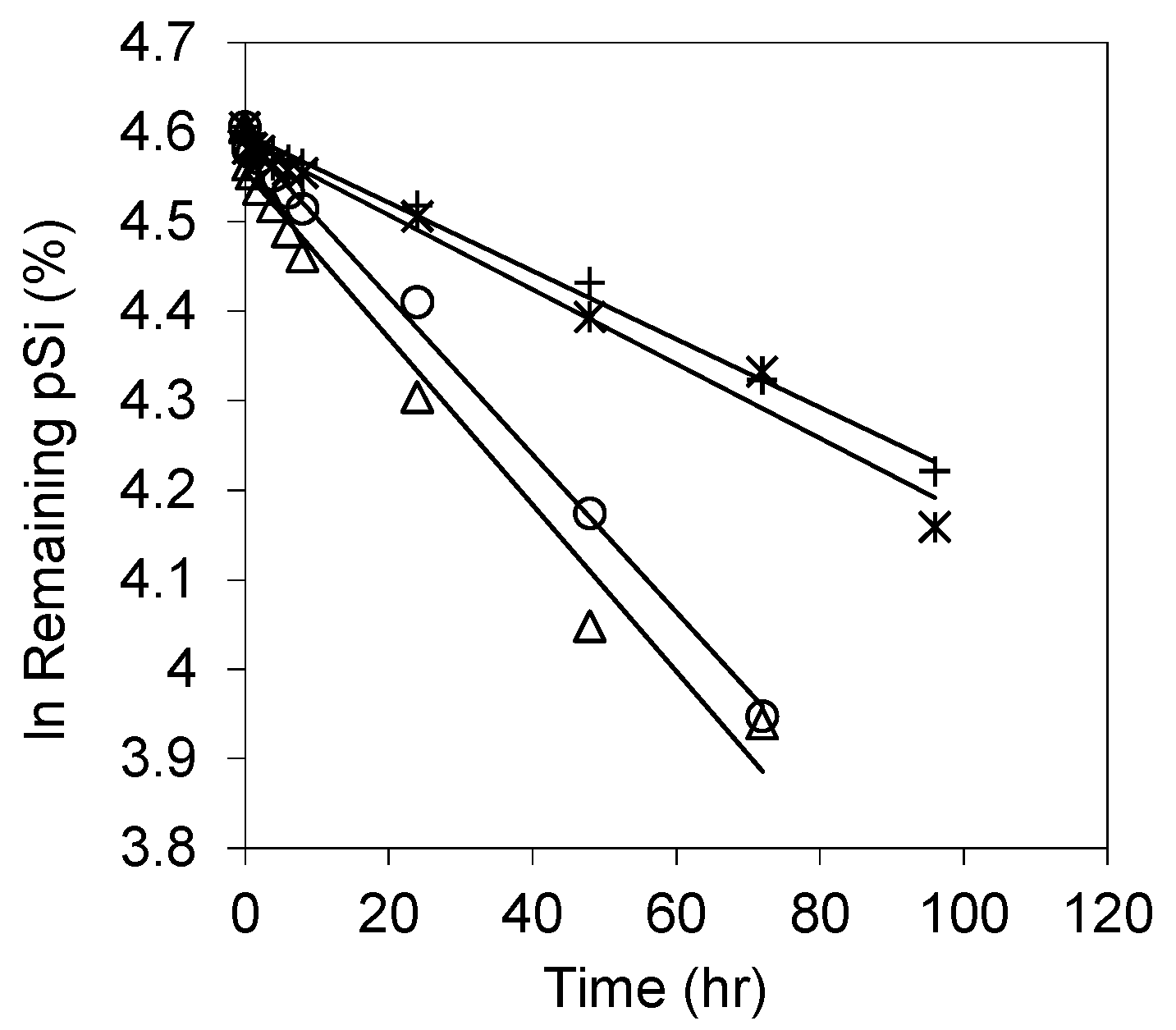
3.3. Influence of Simulated GI Fluids on pSi Dissolution
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Canham, L.T. Bioactive silicon structure fabrication through nanoetching techniques. Adv. Mater. 1995, 7, 1033–1037. [Google Scholar] [CrossRef]
- Korhonen, E.; Rönkkö, S.; Hillebrand, S.; Riikonen, J.; Xu, W.; Järvinen, K.; Lehto, V.-P.; Kauppinen, A. Cytotoxicity assessment of porous silicon microparticles for ocular drug delivery. Eur. J. Pharm. Biopharm. 2016, 100, 1–8. [Google Scholar] [CrossRef]
- Tzur-Balter, A.; Shatsberg, Z.; Beckerman, M.; Segal, E.; Artzi, N. Mechanism of erosion of nanostructured porous silicon drug carriers in neoplastic tissues. Nat. Commun. 2015, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Maniya, N.H.; Patel, S.R.; Murthy, Z.V.P. Development and in vitro evaluation of acyclovir delivery system using nanostructured porous silicon carriers. Chem. Eng. Res. Des. 2015, 104, 551–557. [Google Scholar] [CrossRef]
- Wang, F.; Hui, H.; Barnes, T.J.; Barnett, C.; Prestidge, C.A. Oxidized Mesoporous Silicon Microparticles for Improved Oral Delivery of Poorly Soluble Drugs. Mol. Pharm. 2009, 7, 227–236. [Google Scholar] [CrossRef]
- Qi, S.C.; Zhang, P.; Ma, M.; Yao, M.; Wu, J.; Mäkilä, E.; Salonen, J.; Ruskoaho, H.; Xu, Y.; Santos, H.A.; et al. Cellular Internalization-Induced Aggregation of Porous Silicon Nanoparticles for Ultrasound Imaging and Protein-Mediated Protection of Stem Cells. Small 2019, 15, 13. [Google Scholar] [CrossRef]
- Galushka, V.V.; Belobrovaya, O.Y.; Bratashov, D.N.; Kondrateva, O.Y.; Polyanskaya, V.P.; Sidorov, V.I.; Yagudin, I.T.; Terin, D.V. Gamma-Radiation Monitoring of Luminescent Porous Silicon for Tumor Imaging. Bionanoscience 2018, 8, 818–822. [Google Scholar] [CrossRef]
- Yang, Y.; Su, P.; Tang, Y. Stimuli-Responsive Lanthanide-Based Smart Luminescent Materials for Optical Encoding and Bio-applications. ChemNanoMat 2018, 4, 1097–1120. [Google Scholar] [CrossRef]
- Kumeria, T.; McInnes, S.J.P.; Maher, S.; Santos, A. Porous silicon for drug delivery applications and theranostics: Recent advances, critical review and perspectives. Expert Opin. Drug Deliv. 2017, 14, 1407–1422. [Google Scholar] [CrossRef]
- Barnes, T.J.; Prestidge, C.A. Recent advances in porous silicon-based therapeutic delivery. Ther. Deliv. 2015, 6, 97–100. [Google Scholar] [CrossRef]
- Jakobsson, U.; Makila, E.; Airaksinen, A.J.; Alanen, O.; Etile, A.; Koster, U.; Ranjan, S.; Salonen, J.; Santos, H.A.; Helariutta, K. Porous Silicon as a Platform for Radiation Theranostics Together with a Novel RIB-Based Radiolanthanoid. Contrast Media Mol. Imaging 2019, 2019, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-F.; Sarparanta, M.P.; Mäkilä, E.M.; Hyvönen, M.L.K.; Laakkonen, P.M.; Salonen, J.J.; Hirvonen, J.T.; Airaksinen, A.J.; Santos, H.A. Multifunctional porous silicon nanoparticles for cancer theranostics. Biomaterials 2015, 48, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Prestidge, C.A.; Barnes, T.J. 15-Nanoporous silicon to enhance drug solubility. In Porous Silicon for Biomedical Applications; Santos, H.A., Ed.; Woodhead Publishing: Cambridge, UK, 2014; pp. 356–373. [Google Scholar]
- Merazga, S.; Cheriet, A.; M’Hammedi, K.; Mefoued, A.; Gabouze, N. Investigation of porous silicon thin films for electrochemical hydrogen storage. Int. J. Hydrogen Energy 2019, 44, 9994–10002. [Google Scholar] [CrossRef]
- Tang, C.-H.; Li, W.-J.; Hung, C.-H.; Hsiao, P.-H.; Chen, C.-Y. Highly Porous Silicon Nanowires Made with Solvent-Mediated Wet Chemical Etching and Their Thermoelectric Applications. ChemistrySelect 2017, 2, 10865–10870. [Google Scholar] [CrossRef]
- Xia, B.; Zhang, W.; Shi, J.; Li, J.; Chen, Z.; Zhang, Q. NIR light-triggered gelling in situ of porous silicon nanoparticles/PEGDA hybrid hydrogels for localized combinatorial therapy of cancer cells. J. Appl. Polym. Sci. 2019, 136, 7. [Google Scholar] [CrossRef]
- Kurdyukov, D.A.; Eurov, D.A.; Shmakov, S.V.; Kirilenko, D.A.; Kukushkina, J.A.; Smirnov, A.N.; Yagovkina, M.A.; Klimenko, V.V.; Koniakhin, S.V.; Golubev, V.G. Fabrication of doxorubicin-loaded monodisperse spherical micro-mesoporous silicon particles for enhanced inhibition of cancer cell proliferation. Microporous Mesoporous Mater. 2019, 281, 1–8. [Google Scholar] [CrossRef]
- Kang, R.H.; Lee, S.H.; Kang, S.; Kang, J.; Hur, J.K.; Kim, D. Systematic Degradation Rate Analysis of Surface-Functionalized Porous Silicon Nanoparticles. Materials 2019, 12, 580. [Google Scholar] [CrossRef]
- Li, W.; Liu, Z.H.; Fontana, F.; Ding, Y.P.; Liu, D.F.; Hirvonen, J.T.; Santos, H.A. Tailoring Porous Silicon for Biomedical Applications: From Drug Delivery to Cancer Immunotherapy. Adv. Mater. 2018, 30, 1703740. [Google Scholar] [CrossRef]
- Seo, H.; Kim, D.; Ahn, H.S.; Hwang, S.; Luu, Q.S.; Kim, J.; Lee, S.; Lee, Y. Efficient Conversion Method of Bulk Silicon Powders into Porous Silicon Nanoparticles. Bull. Korean Chem. Soc. 2018, 39, 1455–1458. [Google Scholar] [CrossRef]
- Jarvis, K.L.; Barnes, T.J.; Prestidge, C.A. Surface chemistry of porous silicon and implications for drug encapsulation and delivery applications. Adv. Colloid Interface Sci. 2012, 175, 25–38. [Google Scholar] [CrossRef]
- Jarvis, K.L.; Barnes, T.J.; Prestidge, C.A. Surface chemical modification to control molecular interactions with porous silicon. J. Colloid Interface Sci. 2011, 363, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, K.L.; Barnes, T.J.; Prestidge, C.A. Thermal Oxidation for Controlling Protein Interactions with Porous Silicon. Langmuir 2010, 26, 14316–14322. [Google Scholar] [CrossRef] [PubMed]
- Mäkilä, E.; Bimbo, L.M.; Kaasalainen, M.; Herranz, B.; Airaksinen, A.J.; Heinonen, M.; Kukk, E.; Hirvonen, J.; Santos, H.A.; Salonen, J. Amine Modification of Thermally Carbonized Porous Silicon with Silane Coupling Chemistry. Langmuir 2012, 28, 14045–14054. [Google Scholar] [CrossRef] [PubMed]
- Britcher, L.; Barnes, T.J.; Griesser, H.J.; Prestidge, C.A. PEGylation of porous silicon using click chemistry. Langmuir 2008, 24, 7625–7627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Yoshikawa, C.; Welch, N.G.; Pasic, P.; Thissen, H.; Voelcker, N.H. Spatially Controlled Surface Modification of Porous Silicon for Sustained Drug Delivery Applications. Sci. Rep. 2019, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Ogata, Y.H.; Kato, F.; Tsuboi, T.; Sakka, T. Changes in the environment of hydrogen in porous silicon with thermal Annealing. J. Electrochem. Soc. 1998, 145, 2439–2444. [Google Scholar] [CrossRef]
- Hines, M.A.; Chabal, Y.J.; Harris, T.D.; Harris, A.L. Measuring the structure of etched silicon surfaces with Raman spectroscopy. J. Chem. Phys. 1994, 101, 8055. [Google Scholar] [CrossRef]
- Allongue, P.; Brune, H.; Gerischer, H. In situ STM observations of the etching of n-Si(111) in NaOH solutions. Surf. Sci. 1992, 275, 414–423. [Google Scholar] [CrossRef]
- Campbell, S.A.; Schiffrin, D.J.; Tufton, P.J. Chemical and electrochemical anisotropic dissolution of silicon in ethylenediamine + pyrocatechol + water media. J. Electroanal. Chem. 1993, 344, 211–233. [Google Scholar] [CrossRef]
- Steinem, C.; Janshoff, A.; Lin, V.S.Y.; Volcker, N.H.; Ghadiri, R.M. DNA hybridization-enhanced porous silicon corrosion: Mechanistic investigations and prospect for optical interferometric biosensing. Tetrahedron 2004, 60, 11259–11267. [Google Scholar] [CrossRef]
- Zhu, G.; Liu, J.-T.; Wang, Y.; Zhang, D.; Guo, Y.; Tasciotti, E.; Hu, Z.; Liu, X. In Situ Reductive Synthesis of Structural Supported Gold Nanorods in Porous Silicon Particles for Multifunctional Nanovectors. ACS Appl. Mater. Interfaces 2016, 8, 11881–11891. [Google Scholar] [CrossRef] [PubMed]
- Bateman, J.E.; Eagling, R.D.; Horrocks, B.R.; Houlton, A.; Worrall, D.R. Role for organic molecules in the oxidation of porous silicon. Chem. Commun. 1997, 23, 2275–2276. [Google Scholar] [CrossRef]
- Xu, D.; Sun, L.; Li, H.; Zhang, L.; Guo, G.; Zhao, X.; Gui, L. Hydrolysis and silanization of the hydrosilicon surface of freshly prepared porous silicon by an amine catalytic reaction. New J. Chem. 2003, 27, 300–306. [Google Scholar] [CrossRef]
- Floch, J.; Blain, S.; Birot, D.; Treguer, P. In situ determination of silicic acid in sea water based on FIA and colorimetric dual-wavelength measurements. Anal. Chim. Acta 1998, 377, 157–166. [Google Scholar] [CrossRef]
- Anderson, S.H.C.; Elliott, H.; Wallis, D.J.; Canham, L.T.; Powell, J.J. Dissolution of different forms of partially porous silicon wafers under simulated physiological conditions. Phys. Status Solidi 2003, 197, 331–335. [Google Scholar] [CrossRef]
- Manchon, A.; Alkhraisat, M.H.; Rueda-Rodriguez, C.; Pintado, C.; Prados-Frutos, J.C.; Torres, J.; Cabarcos, E.L. Silicon bioceramic loaded with vancomycin stimulates bone tissue regeneration. J. Biomed. Mater. Res. Part B Appl. Biomater. 2018, 106, 2307–2315. [Google Scholar] [CrossRef]
- Low, S.P.; Voelcker, N.H.; Canham, L.T.; Williams, K.A. The biocompatibility of porous silicon in tissues of the eye. Biomaterials 2009, 30, 2873–2880. [Google Scholar] [CrossRef]
- Bowditch, A.P.; Waters, K.; Gale, H.; Rice, P.; Scott, E.A.M.; Canham, L.T.; Reeves, C.L.; Loni, A.; Cox, T.I. In-vivo assessment of tissue compatibility and calcification of bulk and porous silicon. Mater. Res. Soc. Symp. Proc. 1999, 536, 149. [Google Scholar] [CrossRef]
- Wang, F.; Timothy, J.B.; Clive, A.P. Celecoxib confinement within mesoporous silicon for enhanced oral bioavailability. Mesoporous Biomater. 2013, 1, 1–15. [Google Scholar] [CrossRef]
- Kato, Y.; Ito, T.; Hirak, A. Initial oxidation process of anodized porous silicon with hydrogen atoms chemisorbed on the inner surface Japan. J. Appl. Phys. 1988, 27, L1406. [Google Scholar] [CrossRef]
- Ogata, Y.; Niki, H.; Sakka, T.; Iwasaki, M. Oxidation of porous silicon under water vapor environment. J. Electrochem. Soc. 1995, 142, 1595–1601. [Google Scholar] [CrossRef]
- Bateman, J.E.; Horrocks, B.R.; Houlton, A. Reactions of water and methanol at hydrogen-terminated silicon surfaces studied by transmission FTIR. J. Chem. Soc. Faraday Trans. 1997, 93, 2427–2431. [Google Scholar] [CrossRef]
- Haiss, W.; Raisch, P.; Schirin, D.J.; Bitsch, L.; Nichols, R.J. An FTIR study of the surface chemistry of the dynamic Si(100) surface during etching in alkaline solution. Faraday Discuss 2002, 121, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Chabal, Y.J.; Raghavachari, K. New ordered structure for the H-Saturated Si(100) surface: The (3x1) Phase. Phys. Rev. Lett. 1985, 54, 1055. [Google Scholar] [CrossRef] [PubMed]
- Bellet, D.; Dolino, G.; Ligeon, M.; Blanc, P.; Krisch, M. Studies of coherent and diffuse x-ray scattering by porous silicon. J. Appl.Phys. 1992, 71, 145–149. [Google Scholar] [CrossRef]
- Mawhinney, D.B.; Glass, J.A.; Yates, J.T. FTIR study of the oxidation of porous silicon. J. Phys. Chem. B 1997, 101, 1202–1206. [Google Scholar] [CrossRef]
- Kato, Y.; Ito, T.; Hiraki, A. Low temperature oxidation of crystalline porous silicon. Appl. Surf. Sci. 1989, 41–42, 614–618. [Google Scholar] [CrossRef]
- Graf, D.; Grundner, M.; Schulz, R. Reaction of water with hydrofluoric acid treated silicon(111) and (100) surfaces. J. Vac. Sci.Tech. A 1989, 7, 808–813. [Google Scholar] [CrossRef]
- Stefanov, B.B.; Gurevich, A.B.; Weldon, M.K.; Raghavachari, K.; Chabal, Y.J. Silicon Epoxide: Unexpected Intermediate during Silicon Oxide Formation. Phys. Rev. Lett. 1998, 81, 3908. [Google Scholar] [CrossRef]
- Ogata, Y.H.; Tsuboi, T.; Sakka, T.; Naito, S. Oxidation of porous silicon in dry and wet environments under mild temperature conditions. J. Porous Mater. 2000, 7, 63–66. [Google Scholar] [CrossRef]
- Raghavachari, K. First-Principles quantum chemical investigations of silicon oxidation. In Fundamental Aspect of Silicon Oxidation; Chabal, Y.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2001; pp. 127–141. [Google Scholar]
- Morita, M.; Ohmi, T.; Hasegawa, E.; Kawakami, M.; Suma, K. Control factor of native oxide growth on silicon in air or in ultra-pure water. Appl. Phys. Lett. 1989, 55, 562–564. [Google Scholar] [CrossRef]
- Gole, A.; Sastry, M.; Dash, C.; Rao, M. Encapsulation and biocatalytic activity of the enzyme pepsin in fatty lipid films by selective electrostatic interactions. Chem. Commun. 2000, 4, 297–298. [Google Scholar] [CrossRef]
- Uversky, V.N.; Permyakov, A.E.A. Methods in Protein Structure and Stability Analysis: Conformational Stability, Size, Shape and Surface of Protein Molecules. In Molecular Anatomy and Physiology of Protein; Uversky, V.N., Ed.; Nova Publishers: Hauppauge, NY, USA, 2007; p. 404. [Google Scholar]
- Prestidge, C.A.; Barnes, T.J.; Mierczynska-Vasilev, A.; Skinner, W.; Peddie, F.; Barnett, C. Loading and release of a model protein from porous silicon powders. Phys. Status Solidi A Appl. Mater. Sci. 2007, 204, 3361–3366. [Google Scholar] [CrossRef]
- Foraker, A.B.; Walczak, R.J.; Cohen, M.H.; Boiarski, T.A.; Grove, C.F.; Swaan, P.W. Microfabricated porous silicon particles enhance paracellular delivery of insulin across intestinal Caco-2 cell monolayers. Pharm. Res. 2003, 20, 110–116. [Google Scholar] [CrossRef]
- Karlsson, L.M.; Tengvall, P.; Lundstrom, I.; Arwin, H. Penetration and loading of human serum albumin in porous silicon layers with different pore sizes and thicknesses. J. Colloid Interface Sci. 2003, 266, 40–47. [Google Scholar] [CrossRef]
- Cozzone, P.; Pasero, L.; Marchis-Mouren, G. Characterization of Porcine Pancreatic Isoamylases: Separation and Amino Acid Composition. Biochim. Biophys. Acta 1970, 200, 590. [Google Scholar] [CrossRef]
- Bozso, F.; Avouris, P. Reaction of Si(100) with NH3: Rate-limiting steps and reactivity enhancement via electronic excitation. Phys. Rev. Lett. 1986, 57, 1185. [Google Scholar] [CrossRef]
- Truesdale, V.; Greenwood, J.; Rendell, A. The rate-equation for biogenic silica dissolution in seawater—New hypotheses. Aquat. Geochem. 2005, 11, 319–343. [Google Scholar] [CrossRef]
- Lim, P.K.; Tam, W.K.; Yeung, L.F.; Lam, F.M. Effect of hydrogen on dangling bond in a-Si thin film. J. Phys. Conf. Ser. 2007, 61, 708. [Google Scholar] [CrossRef]
- Trucks, G.W.; Raghavachari, K.; Higashi, G.S.; Chabal, Y.J. Mechanism of HF etching of silicon surfaces: A theoretical understanding of hydrogen passivation. Phys. Rev. Lett. 1990, 65, 504. [Google Scholar] [CrossRef]
- O’Connor, T.L.; Greenberg, S.A. The kinetics for the solution of silica in aqueous solutions. J. Phys. Chem. 1958, 62, 1195–1198. [Google Scholar]
- Bunker, B.C. Molecular mechanisms for corrosion of silica and silicate glasses. J. Non-Cryst. Solid 1994, 179, 300–308. [Google Scholar] [CrossRef]
- Jarvis, K.L.; Barnes, T.J.; Badalyan, A.; Pendleton, P.; Prestidge, C.A. Impact of thermal oxidation on the adsorptive properties and structure of porous silicon particles. J. Phys. Chem. C 2008, 112, 9717–9722. [Google Scholar] [CrossRef]
- Pap, A.E.; Kordas, K.; Toth, G.; Levoska, J.; Uusimaki, A.; Vahakangas, J.; Leppavuori, S.; George, T.F. Thermal oxidation of porous silicon: Study on structure. Appl. Phys. Lett. 2005, 86, 041501. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxidation Temperature (°C) | Label | BET-SSA (m2/g) | Mesopore Volume (mL/g) | Mesopore Diameter (nm) |
|---|---|---|---|---|
| unoxidized pSi | pSi | 325.7 | 0.77 | 10.1 |
| 200 | OXpSi-200 | 312.5 | 0.74 | 10.1 |
| 300 | OXpSi-300 | - | - | - |
| 400 | OXpSi-400 | 247.7 | 0.61 | 9.6 |
| 500 | OXpSi-500 | - | - | - |
| 600 | OXpSi-600 | 243.8 | 0.48 | 9.3 |
| Sample | Unoxidized pSi | OXpSi-200 | ||||
|---|---|---|---|---|---|---|
| Fast | Medium | Slow | Fast | Medium | Slow | |
| Rate constant k (h−1) | 6.82 | 0.19 | 0.0475 | 2.05 | 0.463 | 0.00824 |
| Fraction (P) | 0.690 | 0.155 | 0.155 | 0.340 | 0.240 | 0.420 |
| Time points | 0–10 min | 10–240 min | 360 min–24 h | 0–10 min | 10–60 min | 360 min–48 h |
| R2 | 1.0 * | 0.9673 | 0.9944 | 1.0 * | 0.9951 | 0.9217 |
| Sample | Rate Constant (×10−3 h−1) | t½ (h) | R2 |
|---|---|---|---|
| OXpSi-300 | 9.300 | 74.5 | 0.9778 |
| OXpSi-400 | 8.797 | 78.8 | 0.9967 |
| OXpSi-500 | 4.153 | 166.9 | 0.9823 |
| OXpSi-600 | 3.811 | 181.8 | 0.9951 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Barnes, T.J.; Prestidge, C.A. Controlling and Predicting the Dissolution Kinetics of Thermally Oxidised Mesoporous Silicon Particles: Towards Improved Drug Delivery. Pharmaceutics 2019, 11, 634. https://doi.org/10.3390/pharmaceutics11120634
Wang F, Barnes TJ, Prestidge CA. Controlling and Predicting the Dissolution Kinetics of Thermally Oxidised Mesoporous Silicon Particles: Towards Improved Drug Delivery. Pharmaceutics. 2019; 11(12):634. https://doi.org/10.3390/pharmaceutics11120634
Chicago/Turabian StyleWang, Feng, Timothy J. Barnes, and Clive A. Prestidge. 2019. "Controlling and Predicting the Dissolution Kinetics of Thermally Oxidised Mesoporous Silicon Particles: Towards Improved Drug Delivery" Pharmaceutics 11, no. 12: 634. https://doi.org/10.3390/pharmaceutics11120634