Opportunities for Successful Stabilization of Poor Glass-Forming Drugs: A Stability-Based Comparison of Mesoporous Silica Versus Hot Melt Extrusion Technologies

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Thermodynamic Solubility Determination
2.2.2. Ultra-High Performance Liquid Chromatography (UHPLC)
2.2.3. Preparation of Hot Melt Extrudates
2.2.4. Preparation of API-Loaded Silica Formulations
2.2.5. Storage of Samples for Stability Studies
2.2.6. Powder X-Ray Diffraction (PXRD)
2.2.7. Non-Sink-Mini-Dissolution in FaSSIF
2.2.8. Scanning Electron Microscopy (SEM)
2.2.9. Differential Scanning Calorimetry (DSC)
2.2.10. Solid-State Nuclear Magnetic Resonance (SS-NMR) Spectroscopy
3. Results
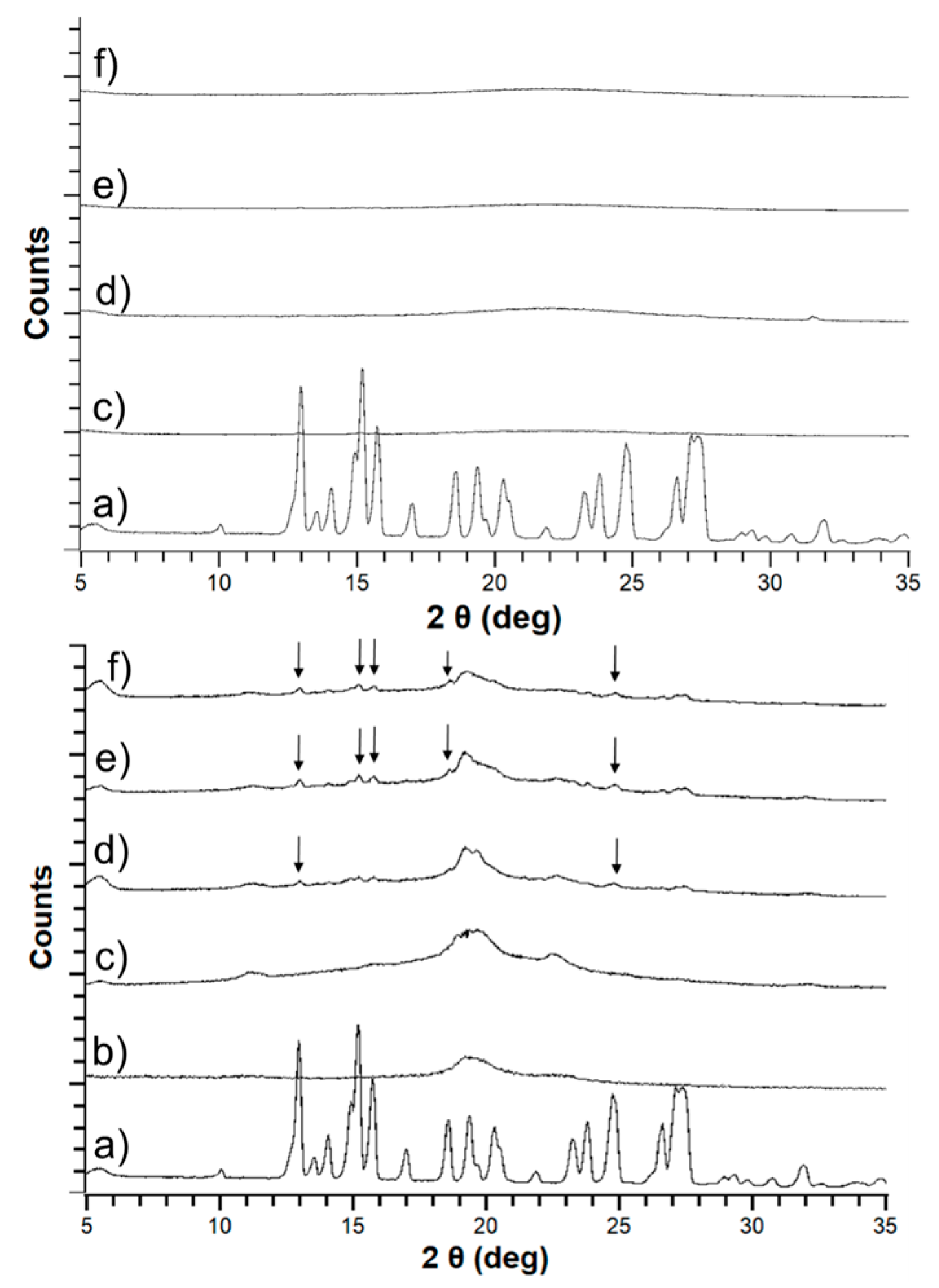
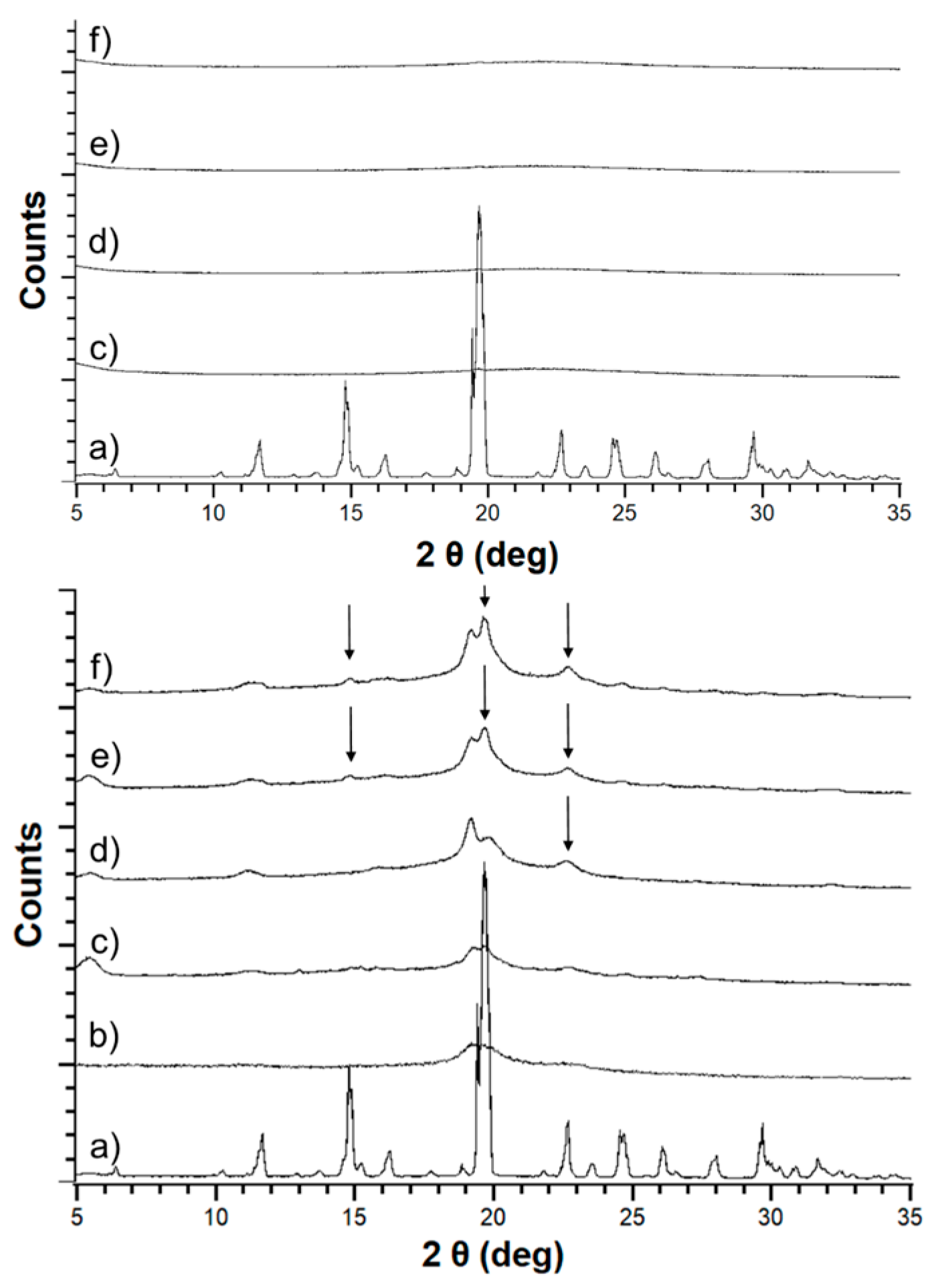
3.1. Macro- and Microscopic Changes
3.2. Solid-State Stability of the Amorphous Form
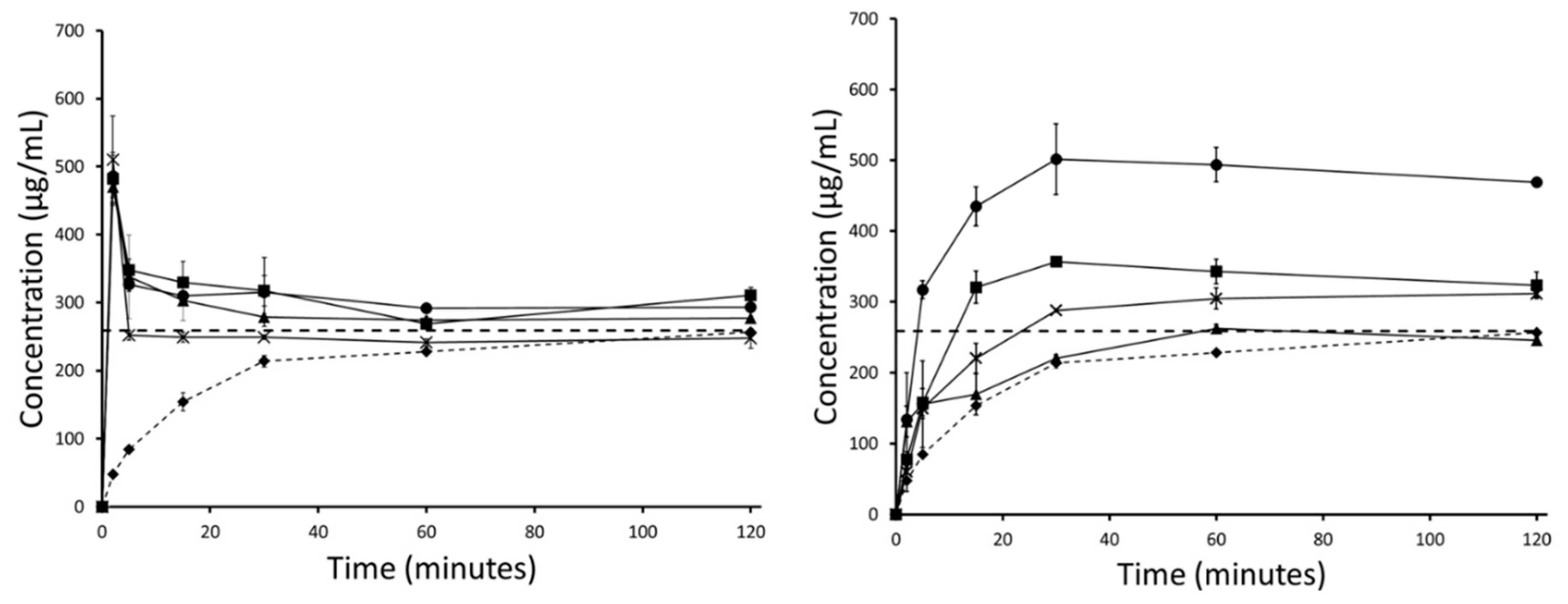
3.3. Stability of the Supersaturated State in FaSSIF
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hancock, B.C.; Parks, M. What is the True Solubility Advantage for Amorphous Pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Rams-Baron, M.; Jachowicz, R.; Boldyreva, E.; Zhou, D.; Jamroz, W.; Paluch, M. Physical Instability: A Key Problem of Amorphous Drugs. In Amorphous Drugs; Springer International Publishing: Cham, Switzerland, 2018; pp. 107–157. [Google Scholar]
- Baird, J.A.; Van Eerdenbrugh, B.; Taylor, L.S. A Classification System to Assess the Crystallization Tendency of Organic Molecules from Undercooled Melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef] [PubMed]
- Alhalaweh, A.; Alzghoul, A.; Mahlin, D.; Bergström, C.A.S. Physical stability of drugs after storage above and below the glass transition temperature: Relationship to glass-forming ability. Int. J. Pharm. 2015, 495, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, N.; Kirchmeyer, W.; Alsenz, J.; Kuentz, M. Theoretical Considerations of the Prigogine–Defay Ratio with Regard to the Glass-Forming Ability of Drugs from Undercooled Melts. Mol. Pharm. 2016, 13, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Van Eerdenbrugh, B.; Baird, J.A.; Taylor, L.S. Crystallization Tendency of Active Pharmaceutical Ingredients Following Rapid Solvent Evaporation—Classification and Comparison with Crystallization Tendency from Under cooled Melts. J. Pharm. Sci. 2010, 99, 3826–3838. [Google Scholar] [CrossRef] [PubMed]
- Ditzinger, F.; Price, D.J.; Ilie, A.-R.; Köhl, N.J.; Jankovic, S.; Tsakiridou, G.; Aleandri, S.; Kalantzi, L.; Holm, R.; Nair, A.; et al. Lipophilicity and hydrophobicity considerations in bio-enabling oral formulations approaches—A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 464–482. [Google Scholar] [CrossRef]
- Blaabjerg, L.I.; Bulduk, B.; Lindenberg, E.; Löbmann, K.; Rades, T.; Grohganz, H. Influence of glass forming ability on the physical stability of supersaturated amorphous solid dispersions. J. Pharm. Sci. 2019, 108, 2561–2569. [Google Scholar] [CrossRef]
- Wyttenbach, N.; Kuentz, M. Glass-forming ability of compounds in marketed amorphous drug products. Eur. J. Pharm. Biopharm. 2017, 112, 204–208. [Google Scholar] [CrossRef]
- Repka, M.A.; Bandari, S.; Kallakunta, V.R.; Vo, A.Q.; McFall, H.; Pimparade, M.B.; Bhagurkar, A.M. Melt extrusion with poorly soluble drugs—An integrated review. Int. J. Pharm. 2018, 535, 68–85. [Google Scholar] [CrossRef]
- Sliwinska-Bartkowiak, M.; Dudziak, G.; Gras, R.; Sikorski, R.; Radhakrishnan, R.; Gubbins, K.E. Freezing behavior in porous glasses and MCM-41. Colloids Surfaces A Physicochem. Eng. Asp. 2001, 187–188, 523–529. [Google Scholar] [CrossRef]
- McCarthy, C.A.; Ahern, R.J.; Dontireddy, R.; Ryan, K.B.; Crean, A.M. Mesoporous silica formulation strategies for drug dissolution enhancement: A review. Expert Opin. Drug Deliv. 2016, 13, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Azaïs, T.; Tourné-Péteilh, C.; Aussenac, F.; Baccile, N.; Coelho, C.; Devoisselle, J.-M.; Babonneau, F. Solid-State NMR Study of Ibuprofen Confined in MCM-41 Material. Chem. Mater. 2006, 18, 6382–6390. [Google Scholar] [CrossRef]
- Müller, R.H.; Wei, Q.; Keck, C.M. CapsMorph: >4 Years Long-Term Stability of Industrially Feasible Amorphous Drug Formulations. In Proceedings of the Annual Meeting of the Controlled Release Society, Honolulu, HI, USA, 21–24 July 2013. [Google Scholar]
- Mellaerts, R.; Houthoofd, K.; Elen, K.; Chen, H.; Van Speybroeck, M.; Van Humbeeck, J.; Augustijns, P.; Mullens, J.; Van den Mooter, G.; Martens, J.A. Aging behavior of pharmaceutical formulations of itraconazole on SBA-15 ordered mesoporous silica carrier material. Microporous Mesoporous Mater. 2010, 130, 154–161. [Google Scholar] [CrossRef]
- Salonen, J.; Kaukonen, A.M.; Hirvonen, J.; Lehto, V.-P. Mesoporous Silicon in Drug Delivery Applications. J. Pharm. Sci. 2008, 97, 632–653. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar]
- Knopp, M.M.; Wendelboe, J.; Holm, R.; Rades, T. Effect of amorphous phase separation and crystallization on the in vitro and in vivo performance of an amorphous solid dispersion. Eur. J. Pharm. Biopharm. 2018, 130, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Guzmán, H.R.; Tawa, M.; Zhang, Z.; Ratanabanangkoon, P.; Shaw, P.; Gardner, C.R.; Chen, H.; Moreau, J.; Almarsson, Ö.; Remenar, J.F. Combined Use of Crystalline Salt Forms and Precipitation Inhibitors to Improve Oral Absorption of Celecoxib from Solid Oral Formulations. J. Pharm. Sci. 2007, 96, 2686–2702. [Google Scholar] [CrossRef]
- Price, D.J.; Ditzinger, F.; Koehl, N.J.; Jankovic, S.; Tsakiridou, G.; Nair, A.; Holm, R.; Kuentz, M.; Dressman, J.B.; Saal, C. Approaches to increase mechanistic understanding and aid in the selection of precipitation inhibitors for supersaturating formulations—A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 483–509. [Google Scholar] [CrossRef]
- Brough, C.; Miller, D.A.; Keen, J.M.; Kucera, S.A.; Lubda, D.; Williams, R.O. Use of Polyvinyl Alcohol as a Solubility-Enhancing Polymer for Poorly Water Soluble Drug Delivery (Part 1). AAPS PharmSciTech 2016, 17, 167–179. [Google Scholar] [CrossRef]
- Price, D.J.; Nair, A.; Kuentz, M.; Dressman, J.; Saal, C. Calculation of drug-polymer mixing enthalpy as a new screening method of precipitation inhibitors for supersaturating pharmaceutical formulations. Eur. J. Pharm. Sci. 2019, 132, 142–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ICH Expert Working Group. ICH Guideline Q1A(R2) Stability Testing of New Drug Substances and Products. International Conference on Harmonization. 2003. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-1-r2-stability-testing-new-drug-substances-products-step-5_en.pdf (accessed on 25 September 2019).
- Vertzoni, M.; Fotaki, N.; Nicolaides, E.; Reppas, C.; Kostewicz, E.; Stippler, E.; Leuner, C.; Dressman, J. Dissolution media simulating the intralumenal composition of the small intestine: Physiological issues and practical aspects. J. Pharm. Pharmacol. 2004, 56, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Dressman, J.B.; Amidon, G.L.; Reppas, C.; Shah, V.P. Dissolution testing as a prognostic tool for oral drug absorption: Immediate release dosage forms. Pharm. Res. 1998, 15, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Edueng, K.; Mahlin, D.; Gråsjö, J.; Nylander, O.; Thakrani, M.; Bergström, C.A.S. Supersaturation Potential of Amorphous Active Pharmaceutical Ingredients after Long-Term Storage. Molecules 2019, 24, 2731. [Google Scholar] [CrossRef] [PubMed]
- Paudel, A.; Geppi, M.; Mooter, G. Van den Structural and Dynamic Properties of Amorphous Solid Dispersions: The Role of Solid-State Nuclear Magnetic Resonance Spectroscopy and Relaxometry. J. Pharm. Sci. 2014, 103, 2635–2662. [Google Scholar] [CrossRef]
- Lee, H.L.; Vasoya, J.M.; Cirqueira, M.D.L.; Yeh, K.L.; Lee, T.; Serajuddin, A.T.M. Continuous Preparation of 1:1 Haloperidol–Maleic Acid Salt by a Novel Solvent-Free Method Using a Twin Screw Melt Extruder. Mol. Pharm. 2017, 14, 1278–1291. [Google Scholar] [CrossRef]
- Djuris, J.; Nikolakakis, I.; Ibric, S.; Djuric, Z.; Kachrimanis, K. Preparation of carbamazepine–Soluplus® solid dispersions by hot-melt extrusion, and prediction of drug–polymer miscibility by thermodynamic model fitting. Eur. J. Pharm. Biopharm. 2013, 84, 228–237. [Google Scholar] [CrossRef]
- Galia, E.; Nicolaides, E.; Hörter, D.; Löbenberg, R.; Reppas, C.; Dressman, J.B. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm. Res. 1998, 15, 698–705. [Google Scholar] [CrossRef]
- Zheng, M.; Bauer, F.; Birk, G.; Lubda, D. Polyvinyl Alcohol in Hot Melt Extrusion to Improve the Solubility of Drugs. Available online: https://www.sigmaaldrich.com/content/dam/sigma-aldrich/0/content/pdf/PS-PVA-HME-Improve-Solubility-03-2017_EN_MS.pdf (accessed on 2 May 2019).
- Forster, A.; Hempenstall, J.; Tucker, I.; Rades, T. Selection of excipients for melt extrusion with two poorly water-soluble drugs by solubility parameter calculation and thermal analysis. Int. J. Pharm. 2001, 226, 147–161. [Google Scholar] [CrossRef]
- Laitinen, R.; Priemel, P.A.; Surwase, S.; Graeser, K.; Strachan, C.J.; Grohganz, H.; Rades, T. Theoretical Considerations in Developing Amorphous Solid Dispersions. In Amorphous Solid Dispersions; Springer: New York, NY, USA, 2014; pp. 35–90. [Google Scholar]
- Niederquell, A.; Wyttenbach, N.; Kuentz, M. New prediction methods for solubility parameters based on molecular sigma profiles using pharmaceutical materials. Int. J. Pharm. 2018, 546, 137–144. [Google Scholar] [CrossRef]
- Loschen, C.; Klamt, A. COSMO quick: A Novel Interface for Fast σ-Profile Composition and Its Application to COSMO-RS Solvent Screening Using Multiple Reference Solvents. Ind. Eng. Chem. Res. 2012, 51, 14303–14308. [Google Scholar] [CrossRef]
- Greenspan, L. Humidity fixed points of binary saturated aqueous solutions. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1977, 81A, 89–96. [Google Scholar] [CrossRef]
- Thiry, J.; Krier, F.; Evrard, B. A review of pharmaceutical extrusion: Critical process parameters and scaling-up. Int. J. Pharm. 2015, 479, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Kissi, E.O.; Ruggiero, M.T.; Hempel, N.-J.; Song, Z.; Grohganz, H.; Rades, T.; Löbmann, K. Characterising glass transition temperatures and glass dynamics in mesoporous silica-based amorphous drugs. Phys. Chem. Chem. Phys. 2019. [Google Scholar] [CrossRef]
- Patel, D.D.; Anderson, B.D. Effect of Precipitation Inhibitors on Indomethacin Supersaturation Maintenance: Mechanisms and Modeling. Mol. Pharm. 2014, 11, 1489–1499. [Google Scholar] [CrossRef]
- Mehta, M.; Suryanarayanan, R. Accelerated Physical Stability Testing of Amorphous Dispersions. Mol. Pharm. 2016, 13, 2661–2666. [Google Scholar] [CrossRef]
- Mehta, M.; Kothari, K.; Ragoonanan, V.; Suryanarayanan, R. Effect of Water on Molecular Mobility and Physical Stability of Amorphous Pharmaceuticals. Mol. Pharm. 2016, 13, 1339–1346. [Google Scholar] [CrossRef]
- Brás, A.R.; Fonseca, I.M.; Dionísio, M.; Schönhals, A.; Affouard, F.; Correia, N.T. Influence of Nanoscale Confinement on the Molecular Mobility of Ibuprofen. J. Phys. Chem. C 2014, 118, 13857–13868. [Google Scholar] [CrossRef]
- Cordeiro, T.; Castiñeira, C.; Mendes, D.; Danède, F.; Sotomayor, J.; Fonseca, I.M.; Gomes da Silva, M.; Paiva, A.; Barreiros, S.; Cardoso, M.M.; et al. Stabilizing Unstable Amorphous Menthol through Inclusion in Mesoporous Silica Hosts. Mol. Pharm. 2017, 14, 3164–3177. [Google Scholar] [CrossRef]
- Kawakami, K. Crystallization Tendency of Pharmaceutical Glasses: Relevance to Compound Properties, Impact of Formulation Process, and Implications for Design of Amorphous Solid Dispersions. Pharmaceutics 2019, 11, 202. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | Flow Rate (mL/min) | % (v/v) Mobile Phase A | % (v/v) Mobile Phase B |
|---|---|---|---|
| 0.00 | 0.83 | 90 | 10 |
| 0.83 | 0.83 | 10 | 90 |
| 1.20 | 1.50 | 90 | 10 |
| 2.00 | 1.50 | 90 | 10 |
| 2.01 | 0.83 | 90 | 10 |
| Formulation | Loading Content (%) (w/w) | |||
|---|---|---|---|---|
| 30 | 20 | 15 | 7.5 | |
| Haloperidol HME | Crystalline | Crystalline | Crystalline | Amorphous |
| Haloperidol loaded silica | Amorphous | Amorphous | Amorphous | Amorphous |
| Carbamazepine HME | Crystalline | Amorphous | Amorphous | Amorphous |
| Carbamazepine loaded silica | Amorphous | Amorphous | Amorphous | Amorphous |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ditzinger, F.; Price, D.J.; Nair, A.; Becker-Baldus, J.; Glaubitz, C.; Dressman, J.B.; Saal, C.; Kuentz, M. Opportunities for Successful Stabilization of Poor Glass-Forming Drugs: A Stability-Based Comparison of Mesoporous Silica Versus Hot Melt Extrusion Technologies. Pharmaceutics 2019, 11, 577. https://doi.org/10.3390/pharmaceutics11110577
Ditzinger F, Price DJ, Nair A, Becker-Baldus J, Glaubitz C, Dressman JB, Saal C, Kuentz M. Opportunities for Successful Stabilization of Poor Glass-Forming Drugs: A Stability-Based Comparison of Mesoporous Silica Versus Hot Melt Extrusion Technologies. Pharmaceutics. 2019; 11(11):577. https://doi.org/10.3390/pharmaceutics11110577
Chicago/Turabian StyleDitzinger, Felix, Daniel J. Price, Anita Nair, Johanna Becker-Baldus, Clemens Glaubitz, Jennifer B. Dressman, Christoph Saal, and Martin Kuentz. 2019. "Opportunities for Successful Stabilization of Poor Glass-Forming Drugs: A Stability-Based Comparison of Mesoporous Silica Versus Hot Melt Extrusion Technologies" Pharmaceutics 11, no. 11: 577. https://doi.org/10.3390/pharmaceutics11110577