
A Comparative Study of Human Pluripotent Stem Cell-Derived Macrophages in Modeling Viral Infections
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Macrophage Differentiation, and Analysis
2.2. HCV Production and Concentration and the Infection of the iMAC-Huh7 Co-Culture
2.3. iMAC Incubation with S. pneumoniae
2.4. Single-Cell RNA-seq of Macrophage Differentiation
2.5. Single-Cell RNA-Seq Data Processing and Analysis
2.6. Bioinformatics Analysis
2.7. Statistical Analysis
2.8. Data Access
3. Results
3.1. Single-Cell Analysis of hPSC-Derived Macrophages
3.2. Gene Expression Change of iMACs upon Encountering Different Human Pathogens
3.3. Cross-Comparison of Transcriptome Changes When iMACs Encounter Different Pathogens
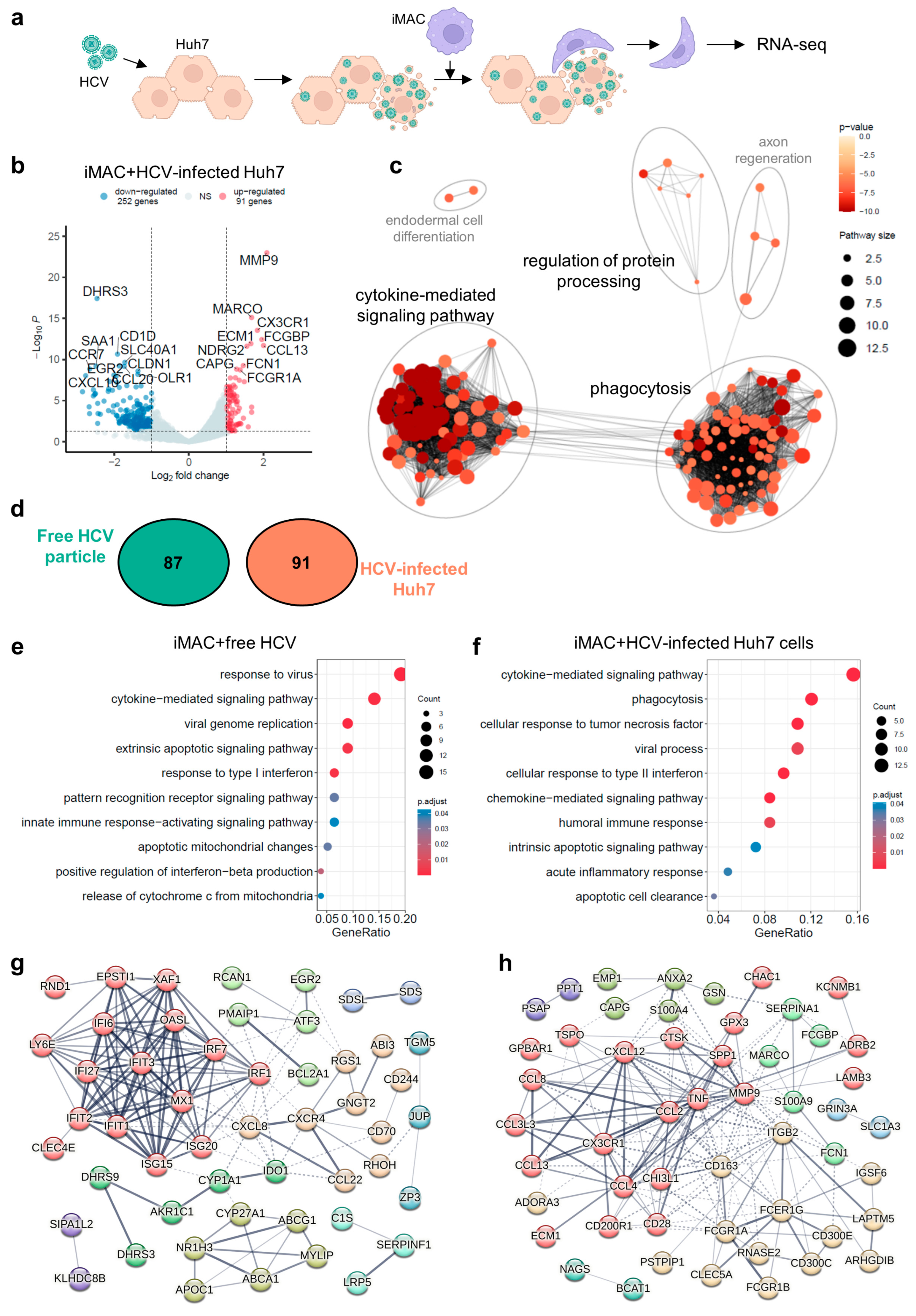
3.4. IMACs Exhibited a Distinct Immune Response upon Free HCV Particles or HCV-Infected Cell Stimulation
3.5. The Presence of iMACs Alleviated the Impact of HCV on Huh7 Cells
3.6. Engineer a High-Content Imaging System to Study Chemical Inhibitor Effects on HCV Infection Using an iMAC-Huh7 Co-Culture System
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boltjes, A.; Movita, D.; Boonstra, A.; Woltman, A.M. The role of Kupffer cells in hepatitis B and hepatitis C virus infections. J. Hepatol. 2014, 61, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Rajab, N.; Rutar, M.; Laslett, A.L.; Wells, C.A. Designer macrophages: Pitfalls and opportunities for modelling macrophage phenotypes from pluripotent stem cells. Differentiation 2018, 104, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Buchrieser, J.; James, W.; Moore, M.D. Human Induced Pluripotent Stem Cell-Derived Macrophages Share Ontogeny with MYB-Independent Tissue-Resident Macrophages. Stem Cell Rep. 2017, 8, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Kempf, H.; Hetzel, M.; Hesse, C.; Hashtchin, A.R.; Brinkert, K.; Schott, J.W.; Haake, K.; Kuhnel, M.P.; Glage, S.; et al. Bioreactor-based mass production of human iPSC-derived macrophages enables immunotherapies against bacterial airway infections. Nat. Commun. 2018, 9, 5088. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Z.W.; Kozaki, T.; Ginhoux, F. Studying tissue macrophages in vitro: Are iPSC-derived cells the answer? Nat. Rev. Immunol. 2018, 18, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Yakala, G.K.; van den Hil, F.E.; Cochrane, A.; Mummery, C.L.; Orlova, V.V. Differentiation and Functional Comparison of Monocytes and Macrophages from hiPSCs with Peripheral Blood Derivatives. Stem Cell Rep. 2019, 12, 1282–1297. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, K.R.; Cowley, S.; Martinez, F.O.; Shaw, M.; Minger, S.L.; James, W. Homogeneous monocytes and macrophages from human embryonic stem cells following coculture-free differentiation in M-CSF and IL-3. Exp. Hematol. 2008, 36, 1167–1175. [Google Scholar] [CrossRef]
- Takata, K.; Kozaki, T.; Lee, C.Z.W.; Thion, M.S.; Otsuka, M.; Lim, S.; Utami, K.H.; Fidan, K.; Park, D.S.; Malleret, B.; et al. Induced-Pluripotent-Stem-Cell-Derived Primitive Macrophages Provide a Platform for Modeling Tissue-Resident Macrophage Differentiation and Function. Immunity 2017, 47, 183–198 e6. [Google Scholar] [CrossRef]
- Dietz, C.; Maasoumy, B. Direct-Acting Antiviral Agents for Hepatitis C Virus Infection-From Drug Discovery to Successful Implementation in Clinical Practice. Viruses 2022, 14, 1325. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.H. 25 years of interferon-based treatment of chronic hepatitis C: An epoch coming to an end. Nat. Rev. Immunol. 2013, 13, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, Y.; Xiong, L.; Li, Y.; Zhang, Y.; Zhao, J.; Jiang, H.; Li, C.; Liu, Y.; Liu, X.; et al. CD127 imprints functional heterogeneity to diversify monocyte responses in inflammatory diseases. J. Exp. Med. 2022, 219, e20211191. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Huang, R.; Zhang, F.; Zhu, Y.; Wang, L.; Chen, X.; Bai, L.; Guo, W.; Chang, S.C.; Hu, X.; et al. Biphasic modulation of insulin signaling enables highly efficient hematopoietic differentiation from human pluripotent stem cells. Stem Cell Res. Ther. 2018, 9, 205. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Duan, F.; Qiao, H.; Gong, M.; Qiu, H.; Chen, X.; Wang, P.; He, Y.; Ding, Q.; et al. Modeling antiviral response in the liver using human pluripotent stem cell-derived macrophages. Life Med. 2024, 3, lnae001. [Google Scholar] [CrossRef]
- Wang, L.; Xu, X.; Cao, Y.; Li, Z.; Cheng, H.; Zhu, G.; Duan, F.; Na, J.; Han, J.J.; Chen, Y.G. Activin/Smad2-induced Histone H3 Lys-27 Trimethylation (H3K27me3) Reduction Is Crucial to Initiate Mesendoderm Differentiation of Human Embryonic Stem Cells. J. Biol. Chem. 2017, 292, 1339–1350. [Google Scholar] [CrossRef]
- Zhang, F.; Zhu, Y.; Chen, J.; Kuang, W.; Huang, R.; Duan, F.; Li, Y.; Wang, L.; Qiu, H.; Chen, X.; et al. Efficient endothelial and smooth muscle cell differentiation from human pluripotent stem cells through a simplified insulin-free culture system. Biomaterials 2021, 271, 120713. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 2024, 42, 293–304. [Google Scholar] [CrossRef]
- Germain, P.L.; Lun, A.; Garcia Meixide, C.; Macnair, W.; Robinson, M.D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res 2021, 10, 979. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Kerseviciute, I.; Gordevicius, J. aPEAR: An R package for autonomous visualisation of pathway enrichment networks. Bioinformatics 2023, 39, btad672. [Google Scholar] [CrossRef]
- Bian, Z.; Gong, Y.; Huang, T.; Lee, C.Z.W.; Bian, L.; Bai, Z.; Shi, H.; Zeng, Y.; Liu, C.; He, J.; et al. Deciphering human macrophage development at single-cell resolution. Nature 2020, 582, 571–576. [Google Scholar] [CrossRef]
- Lian, Q.; Zhang, K.; Zhang, Z.; Duan, F.; Guo, L.; Luo, W.; Mok, B.W.; Thakur, A.; Ke, X.; Motallebnejad, P.; et al. Differential effects of macrophage subtypes on SARS-CoV-2 infection in a human pluripotent stem cell-derived model. Nat. Commun. 2022, 13, 2028. [Google Scholar] [CrossRef] [PubMed]
- Cui, A.; Li, B.; Wallace, M.S.; Gonye, A.L.K.; Oetheimer, C.; Patel, H.; Tonnerre, P.; Holmes, J.A.; Lieb, D.; Yao, B.S.; et al. Single-cell atlas of the liver myeloid compartment before and after cure of chronic viral hepatitis. J. Hepatol. 2024, 80, 251–267. [Google Scholar] [CrossRef]
- An, H.; Qian, C.; Huang, Y.; Li, J.; Tian, X.; Feng, J.; Hu, J.; Fang, Y.; Jiao, F.; Zeng, Y.; et al. Functional vulnerability of liver macrophages to capsules defines virulence of blood-borne bacteria. J. Exp. Med. 2022, 219, e20212032. [Google Scholar] [CrossRef]
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef]
- Fensterl, V.; Sen, G.C. Interferon-induced Ifit proteins: Their role in viral pathogenesis. J. Virol. 2015, 89, 2462–2468. [Google Scholar] [CrossRef]
- Xie, Z.; Chan, E.C.; Druey, K.M. R4 Regulator of G Protein Signaling (RGS) Proteins in Inflammation and Immunity. AAPS J. 2016, 18, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. How macrophages deal with death. Nat. Rev. Immunol. 2019, 19, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Jennelle, L.T.; Magoro, T.; Angelucci, A.R.; Dandekar, A.; Hahn, Y.S. Hepatitis C Virus Alters Macrophage Cholesterol Metabolism Through Interaction with Scavenger Receptors. Viral Immunol. 2022, 35, 223–235. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Qiu, H.; Duan, F.; An, H.; Qiao, H.; Zhang, X.; Zhang, J.-R.; Ding, Q.; Na, J. A Comparative Study of Human Pluripotent Stem Cell-Derived Macrophages in Modeling Viral Infections. Viruses 2024, 16, 552. https://doi.org/10.3390/v16040552
Zhang Y, Qiu H, Duan F, An H, Qiao H, Zhang X, Zhang J-R, Ding Q, Na J. A Comparative Study of Human Pluripotent Stem Cell-Derived Macrophages in Modeling Viral Infections. Viruses. 2024; 16(4):552. https://doi.org/10.3390/v16040552
Chicago/Turabian StyleZhang, Yaxuan, Hui Qiu, Fuyu Duan, Haoran An, Huimin Qiao, Xingwu Zhang, Jing-Ren Zhang, Qiang Ding, and Jie Na. 2024. "A Comparative Study of Human Pluripotent Stem Cell-Derived Macrophages in Modeling Viral Infections" Viruses 16, no. 4: 552. https://doi.org/10.3390/v16040552