
Genomic Characterization of a Bataï Orthobunyavirus, Previously Classified as Ilesha Virus, from Field-Caught Mosquitoes in Senegal, Bandia 1969

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. The Virus
2.2. Sequencing
2.3. Genetics Analyses and Phylogeny
3. Results
3.1. Blast
3.2. Genetic Distance
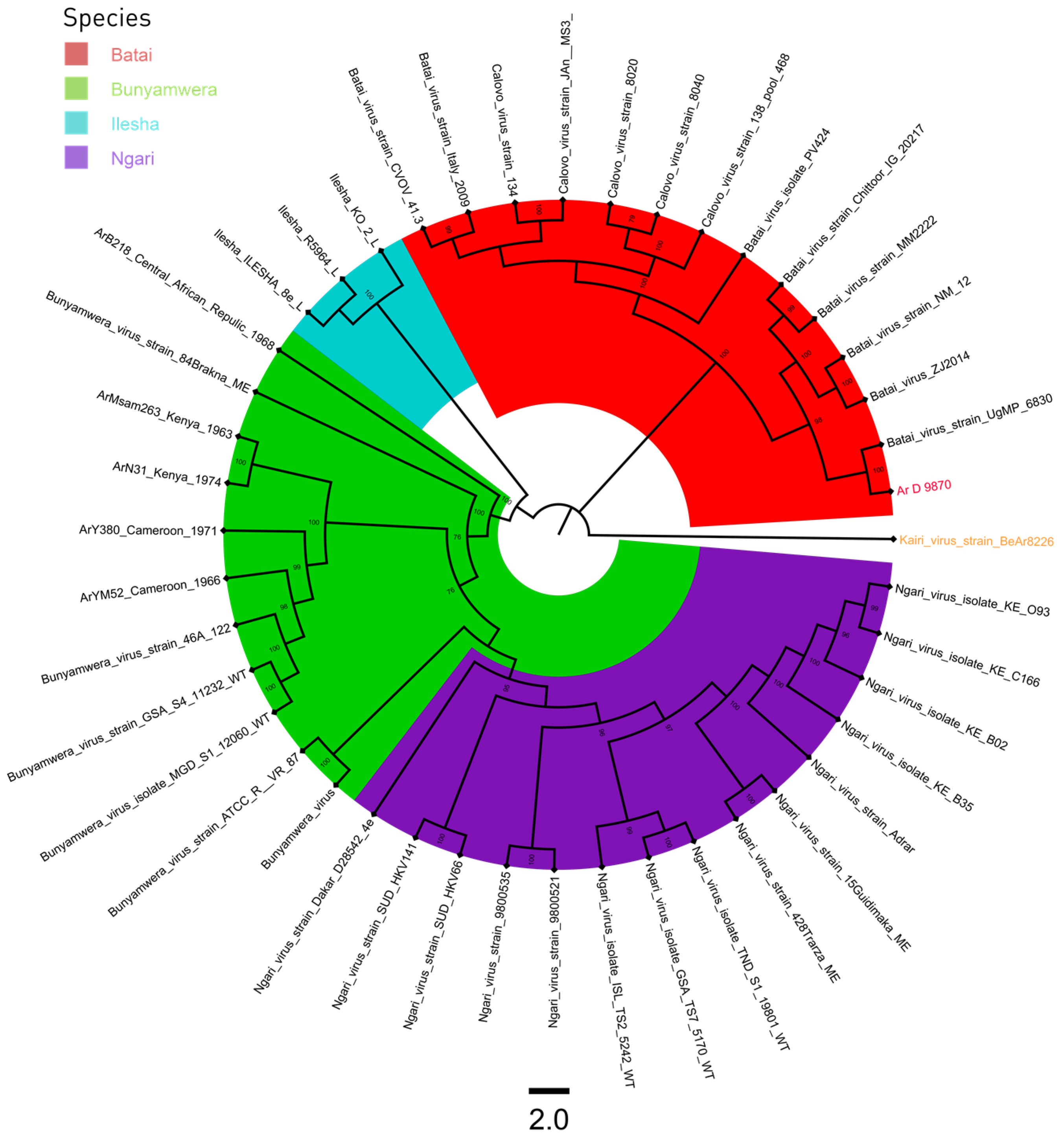
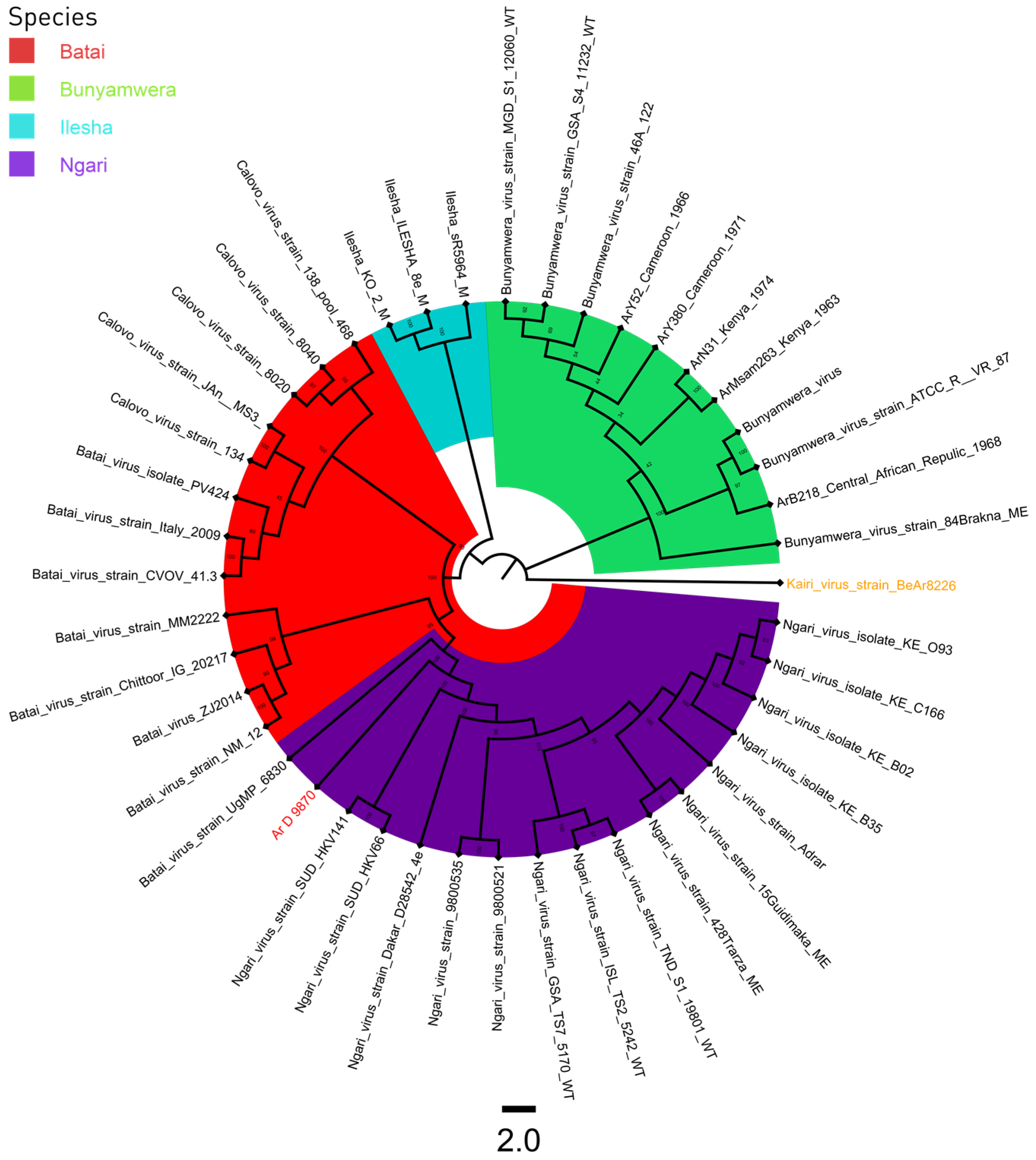
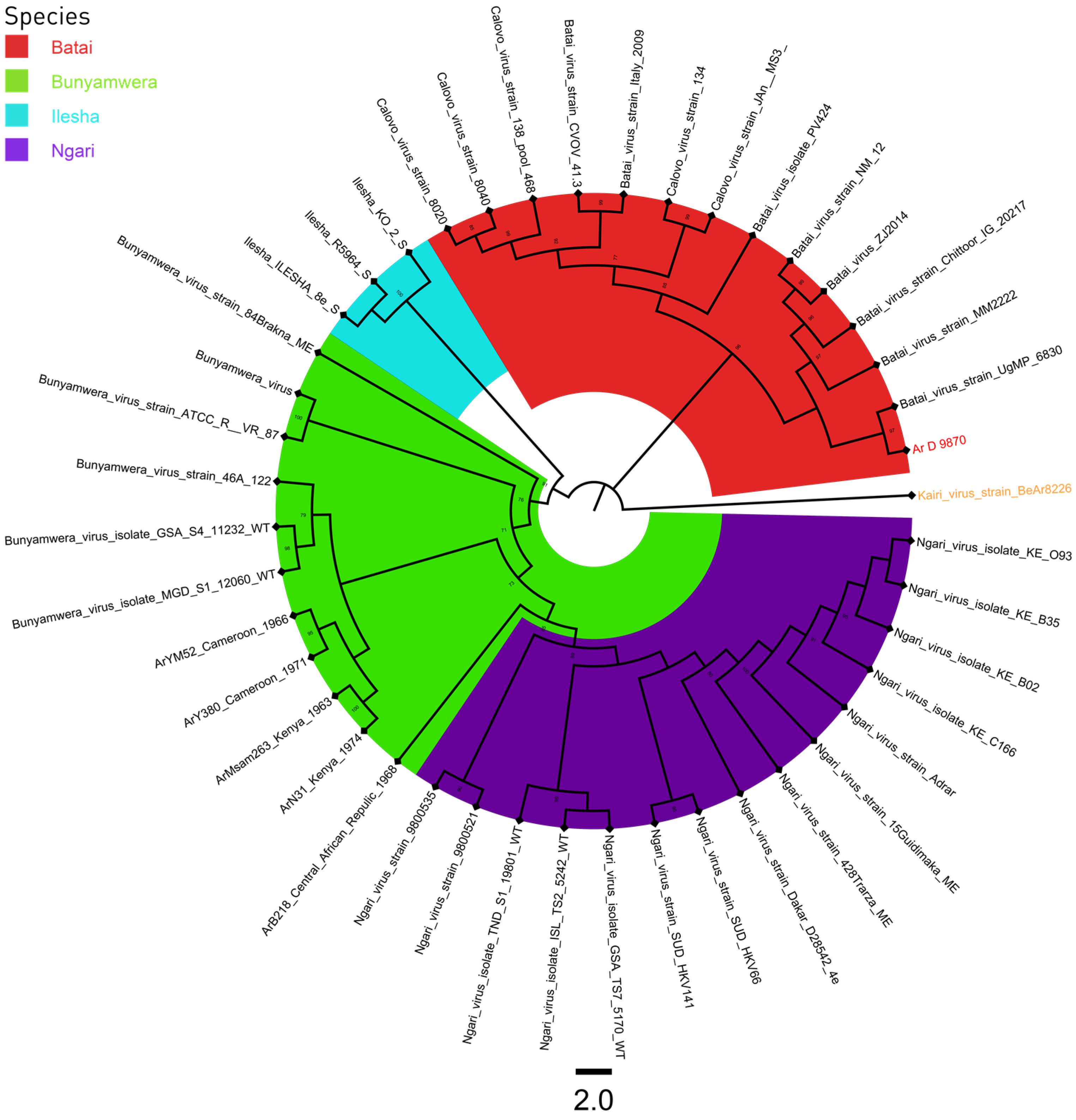
3.3. Phylogeny
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hughes, H.R.; Adkins, S.; Alkhovskiy, S.; Beer, M.; Blair, C.; Calisher, C.H.; Drebot, M.; Lambert, A.J.; de Souza, W.M.; Marklewitz, M.; et al. ICTV Virus Taxonomy Profile: Peribunyaviridae. J. Gen. Virol. 2020, 101, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Hubálek, Z. Mosquito-Borne Viruses in Europe. Parasitol. Res. 2008, 103, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Dilcher, M.; Sall, A.A.; Hufert, F.T.; Weidmann, M. Clarifying Bunyamwera Virus Riddles of the Past. Virus Genes 2013, 47, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Dutuze, M.F.; Nzayirambaho, M.; Mores, C.N.; Christofferson, R.C. A Review of Bunyamwera, Batai, and Ngari Viruses: Understudied Orthobunyaviruses With Potential One Health Implications. Front. Vet. Sci. 2018, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Briese, T.; Bird, B.; Kapoor, V.; Nichol, S.T.; Lipkin, W.I. Batai and Ngari Viruses: M Segment Reassortment and Association with Severe Febrile Disease Outbreaks in East Africa. J. Virol. 2006, 80, 5627–5630. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, K.L.; Folly, A.J.; Hernández-Triana, L.M.; Sewgobind, S.; Johnson, N. Batai Orthobunyavirus: An Emerging Mosquito-Borne Virus in Europe. Viruses 2022, 14, 1868. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.M. Orthobunyaviruses: Recent Genetic and Structural Insights. Nat. Rev. Microbiol. 2014, 12, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Current ICTV Taxonomy Release|ICTV. Available online: https://ictv.global/taxonomy (accessed on 26 April 2023).
- Contigiani, M.S.; Diaz, L.A.; Tauro, L.B. Bunyaviruses. In Arthropod Borne Diseases; Marcondes, C.B., Ed.; Springer International Publishing: New York City, NY, USA, 2017; pp. 137–154. ISBN 978-3-319-13883-1. [Google Scholar]
- Pachler, K.; Růžek, D.; Nowotny, N. Molecular Characterization of the African Orthobunyavirus Ilesha Virus. Infect. Genet. Evol. 2013, 20, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Morvan, J.M.; Digoutte, J.P.; Marsan, P.; Roux, J.F. Ilesha Virus: A New Aetiological Agent of Haemorrhagic Fever in Madagascar. Trans. R. Soc. Trop. Med. Hyg. 1994, 88, 205. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.R.; Rosenthal, K.; Pfaller, M.A. Medical Microbiology-ClinicalKey, 9th ed.; Elsevier: Amsterdam, The Netherlands, 2020; ISBN 978-0-323-67322-8. [Google Scholar]
- Bob, N.S.; Barry, M.A.; Diagne, M.M.; Faye, M.; Ndione, M.H.D.; Diallo, A.; Diop, M.; Diop, B.; Faye, O.; Loucoubar, C.; et al. Detection of Rift Valley Fever Virus Lineage H From South Africa Through the Syndromic Sentinel Surveillance Network in Senegal. Open Forum Infect. Dis. 2022, 9, ofab655. [Google Scholar] [CrossRef] [PubMed]
- Kalantar, K.L.; Carvalho, T.; de Bourcy, C.F.A.; Dimitrov, B.; Dingle, G.; Egger, R.; Han, J.; Holmes, O.B.; Juan, Y.-F.; King, R.; et al. IDseq-An Open Source Cloud-Based Pipeline and Analysis Service for Metagenomic Pathogen Detection and Monitoring. Gigascience 2020, 9, giaa111. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, V. Rambaut’s Full-Sized Avatar A. FigTree 2018. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 18 October 2023).
- Ishihara, Y.; Shioda, C.; Bangphoomi, N.; Sugiura, K.; Saeki, K.; Tsuda, S.; Iwanaga, T.; Takenaka-Uema, A.; Kato, K.; Murakami, S.; et al. Akabane Virus Nonstructural Protein NSm Regulates Viral Growth and Pathogenicity in a Mouse Model. J. Vet. Med. Sci. 2016, 78, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Horne, K.M.; Vanlandingham, D.L. Bunyavirus-Vector Interactions. Viruses 2014, 6, 4373–4397. [Google Scholar] [CrossRef] [PubMed]
- Gerrard, S.R.; Li, L.; Barrett, A.D.; Nichol, S.T. Ngari Virus Is a Bunyamwera Virus Reassortant That Can Be Associated with Large Outbreaks of Hemorrhagic Fever in Africa. J. Virol. 2004, 78, 8922–8926. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Segment L | Segment M | Segment S | |
|---|---|---|---|
| Mapped Reads | 23,210 | 17,746 | 1667 |
| GC Content | 33.9% | 36.4% | 40.6% |
| SNPs | 241 | 148 | 14 |
| %id | 96.5% | 96.6% | 98.5% |
| Informative Nucleotides | 6870 | 4338 | 927 |
| % Genome Called | 100% | 98.5% | 98.1% |
| Missing Bases | 0 | 63 | 16 |
| Ambiguous Bases | 0 | 0 | 0 |
| Reference length | 6870 | 4404 | 945 |
| Coverage Depth | 403.9× | 477.5× | 191.7× |
| Coverage Breadth | 100% | 99.9% | 100% |
| Reference Sequence | JX846603.1-Bataï virus strain UgMP-6830 segment L, complete sequence | DQ436460.1-Bataï virus strain UgMP-6830 segment M polyprotein gene, complete cds | JX846601.1-Bataï virus strain UgMP-6830 segment S, complete sequence |
| AR D 9870 | Bataï | UgMP-6830 | Bunv | Ngari | |
|---|---|---|---|---|---|
| S segment | |||||
| AR D 9870 | 1.375 | 0.000 | 3.887 | 3.797 | |
| BATAÏ | 3.167 | 1.375 | 3.833 | 3.759 | |
| UGMP-6830 | 0.000 | 3.167 | 3.887 | 3.797 | |
| BUNV | 16.909 | 17.348 | 16.909 | 0.599 | |
| NGARI | 16.267 | 16.767 | 16.267 | 1.170 | |
| M segment | |||||
| AR D 9870 | 8.539 | 5.073 | 16.775 | 4.313 | |
| BATAÏ | 125.667 | 8.151 | 16.128 | 8.384 | |
| UGMP-6830 | 25.000 | 117.667 | 16.734 | 5.471 | |
| BUNV | 484.909 | 486.159 | 485.273 | 16.751 | |
| NGARI | 30.733 | 131.533 | 42.467 | 492.103 | |
| L segment | |||||
| AR D 9870 | 11.491 | 7.586 | 20.035 | 20.040 | |
| BATAÏ | 221.000 | 11.907 | 19.265 | 19.324 | |
| UGMP-6830 | 59.000 | 233.250 | 19.934 | 19.856 | |
| BUNV | 609.636 | 611.689 | 610.727 | 6.607 | |
| NGARI | 608.667 | 614.711 | 608.333 | 91.867 | |
| Bataï_Africa | Bataï_Asia | Bataï_Europe | |
|---|---|---|---|
| S segment | |||
| Bataï_Africa | 1.030 | 2.001 | |
| Bataï_Asia | 1.250 | 2.285 | |
| Bataï_Europe | 4.143 | 5.393 | |
| M segment | |||
| Bataï_Africa | 8.587 | 11.173 | |
| Bataï_Asia | 103.375 | 10.986 | |
| Bataï_Europe | 144.214 | 148.679 | |
| L segment | |||
| Bataï_Africa | 11.046 | 14.762 | |
| Bataï_Asia | 173.750 | 14.726 | |
| Bataï_Europe | 275.571 | 290.536 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toure, C.T.; Dieng, I.; Sankhe, S.; Kane, M.; Dia, M.; Mhamadi, M.; Ndiaye, M.; Faye, O.; Sall, A.A.; Diagne, M.M.; et al. Genomic Characterization of a Bataï Orthobunyavirus, Previously Classified as Ilesha Virus, from Field-Caught Mosquitoes in Senegal, Bandia 1969. Viruses 2024, 16, 261. https://doi.org/10.3390/v16020261
Toure CT, Dieng I, Sankhe S, Kane M, Dia M, Mhamadi M, Ndiaye M, Faye O, Sall AA, Diagne MM, et al. Genomic Characterization of a Bataï Orthobunyavirus, Previously Classified as Ilesha Virus, from Field-Caught Mosquitoes in Senegal, Bandia 1969. Viruses. 2024; 16(2):261. https://doi.org/10.3390/v16020261
Chicago/Turabian StyleToure, Cheikh Talibouya, Idrissa Dieng, Safietou Sankhe, Mouhamed Kane, Moussa Dia, Moufid Mhamadi, Mignane Ndiaye, Ousmane Faye, Amadou Alpha Sall, Moussa Moise Diagne, and et al. 2024. "Genomic Characterization of a Bataï Orthobunyavirus, Previously Classified as Ilesha Virus, from Field-Caught Mosquitoes in Senegal, Bandia 1969" Viruses 16, no. 2: 261. https://doi.org/10.3390/v16020261