
Genomic Expedition: Deciphering Human Adenovirus Strains from the 2023 Outbreak in West Bengal, India: Insights into Viral Evolution and Molecular Epidemiology
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Source
2.2. Real-Time PCR of Respiratory Viral Panel
2.3. Amplicon Preparation for Ion Torrent NGS Platform (Ion GeneStudio S5 System)
2.4. Library Preparation for the Ion Torrent NGS Platform (Ion GeneStudio S5 System)
2.5. Sanger Sequencing of the 5′ ITR End and 3′ ITR End
2.6. Bioinformatic Analysis
3. Results
3.1. Genetic Characterisation of Adenovirus-Positive Clinical Samples Using Whole Genome Sequence Analysis
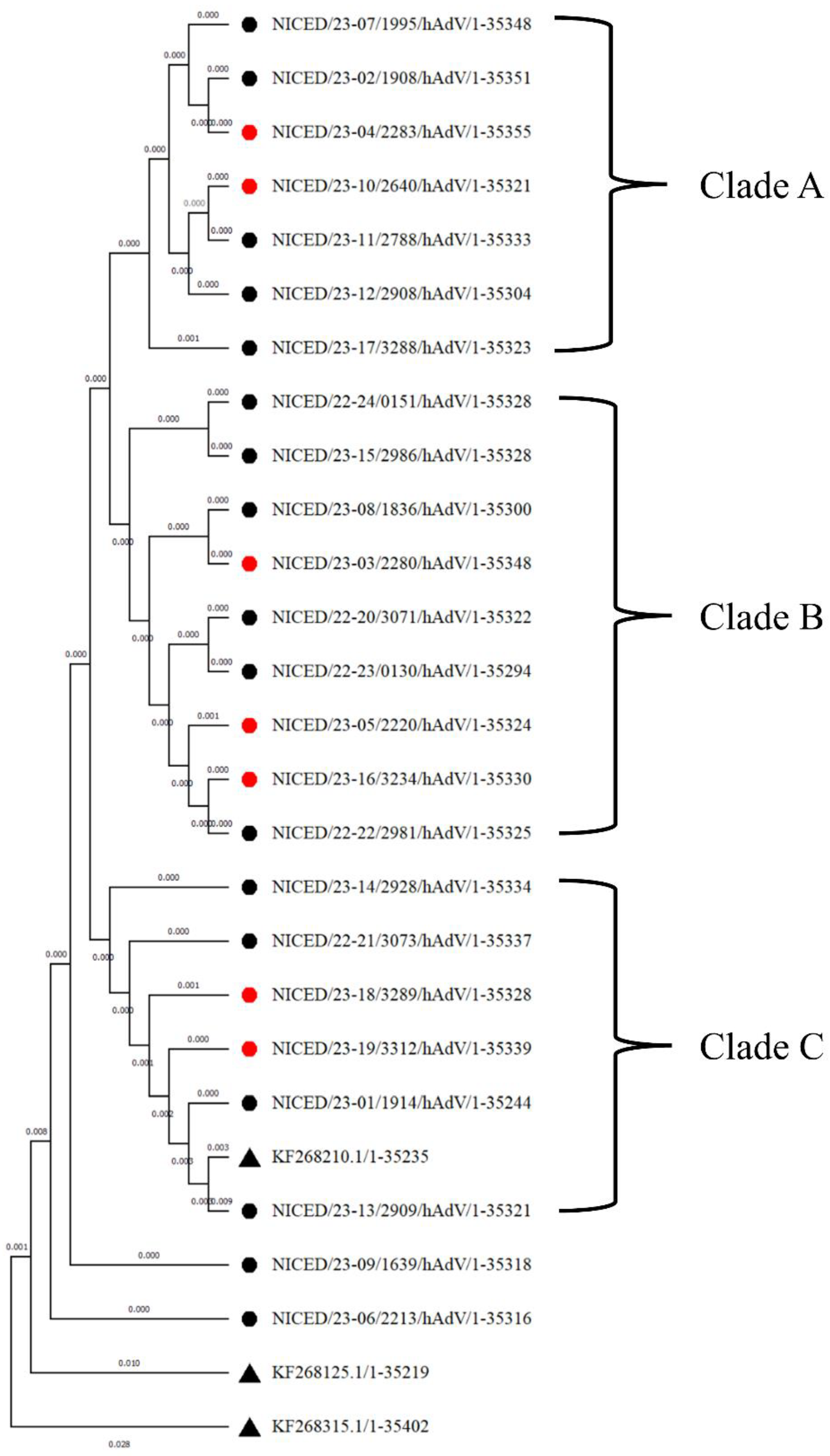
3.2. Phylogenetic Analysis
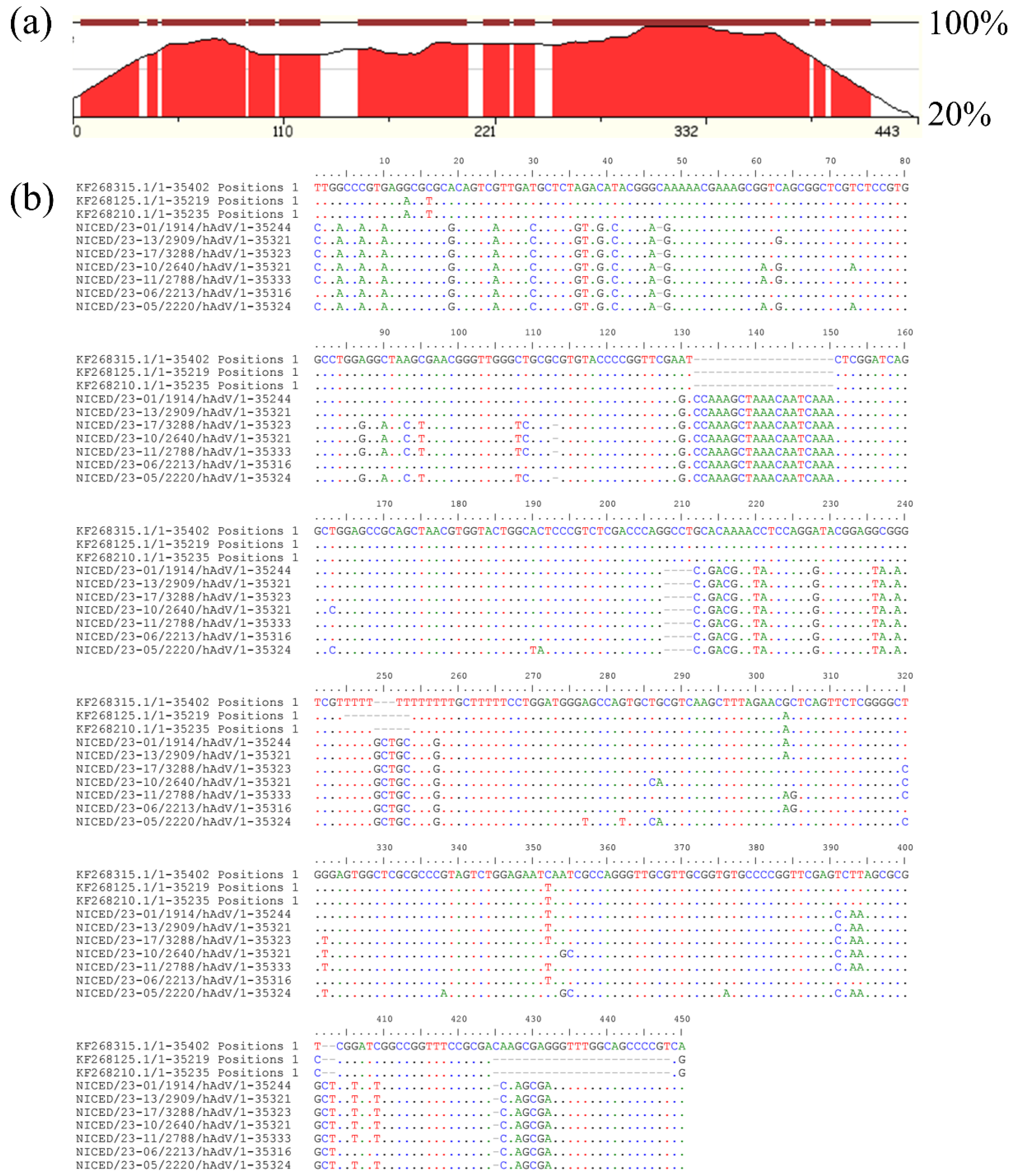
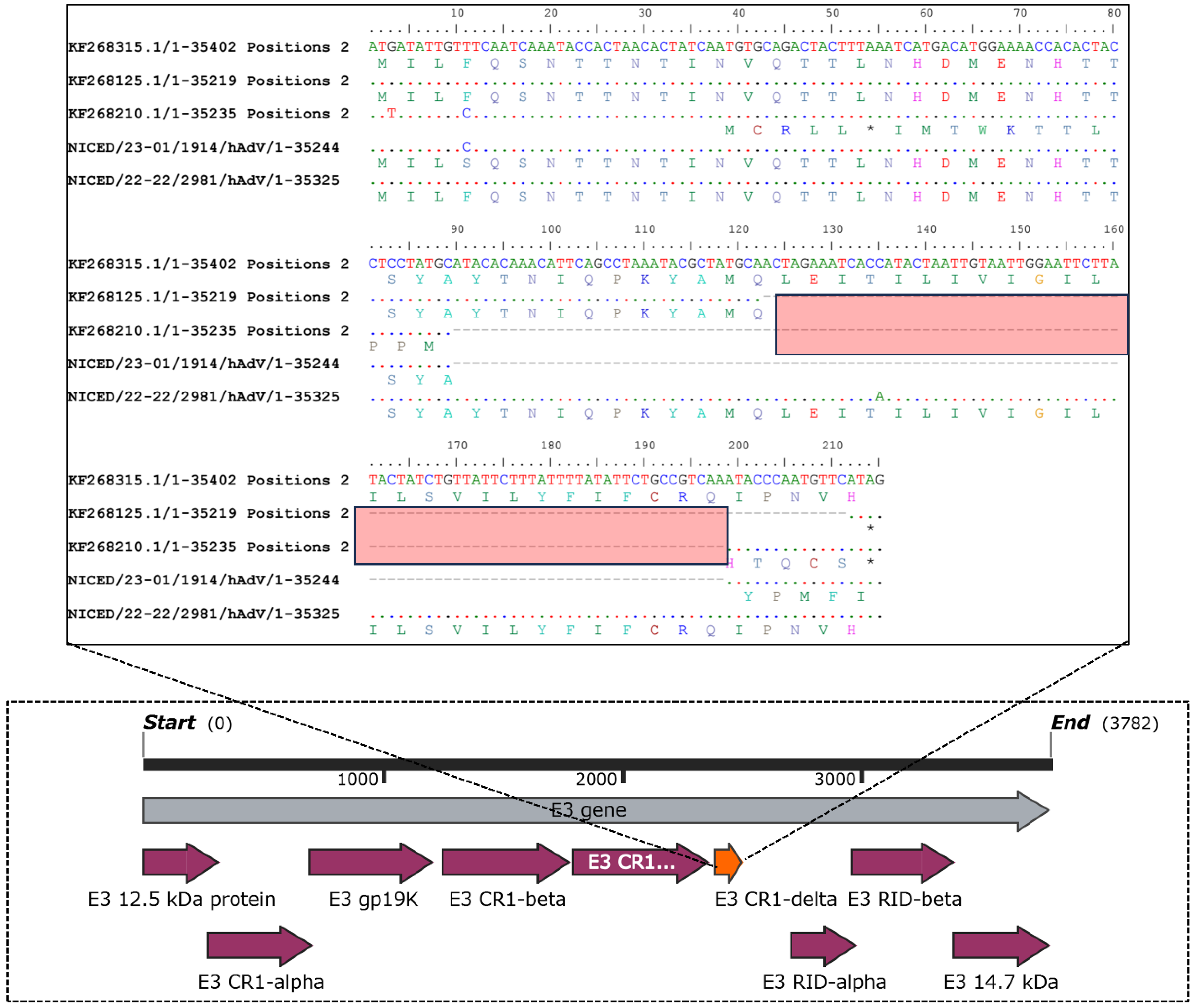
3.3. Inspection of Multiple Sequence Alignment (MSA)
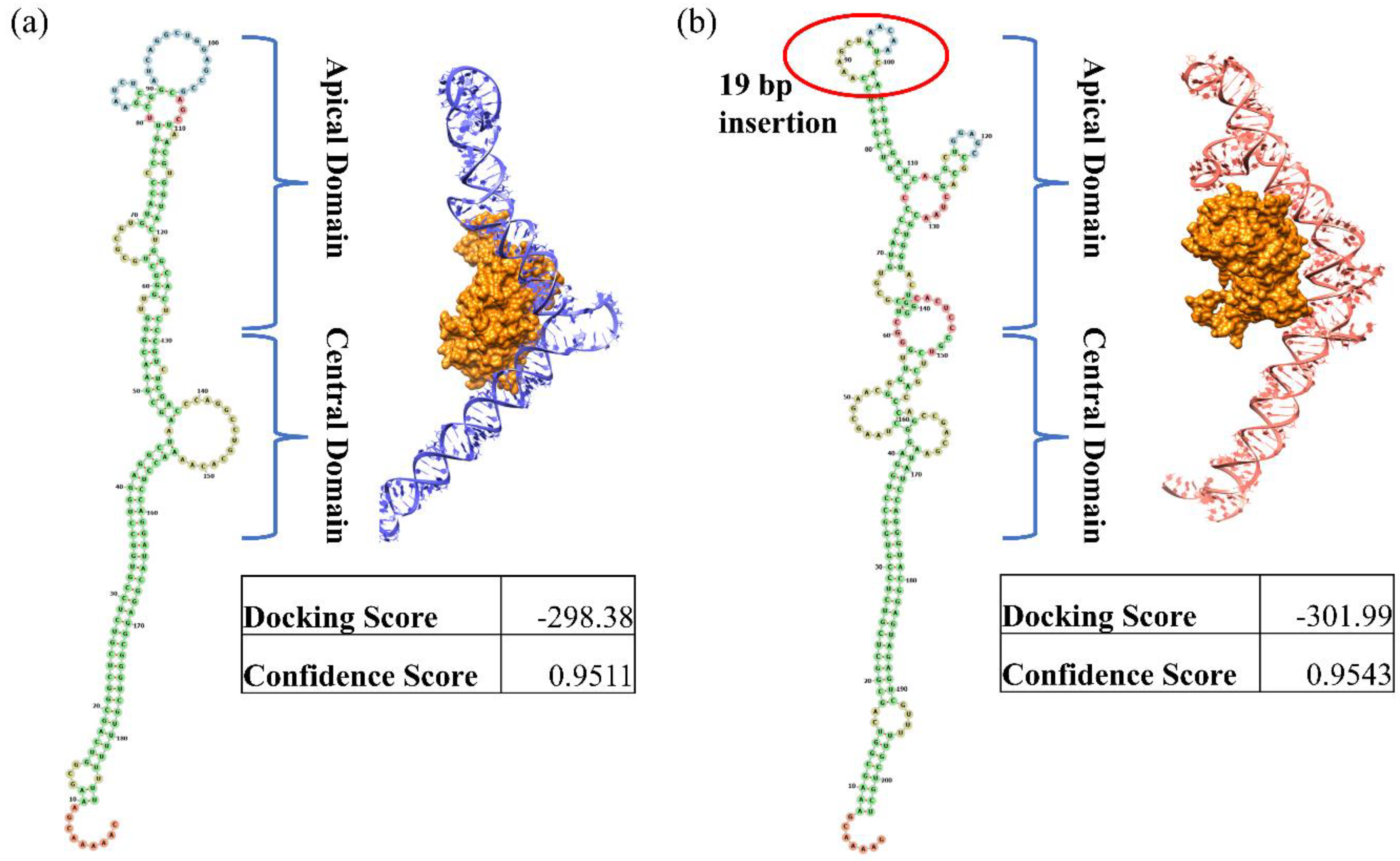
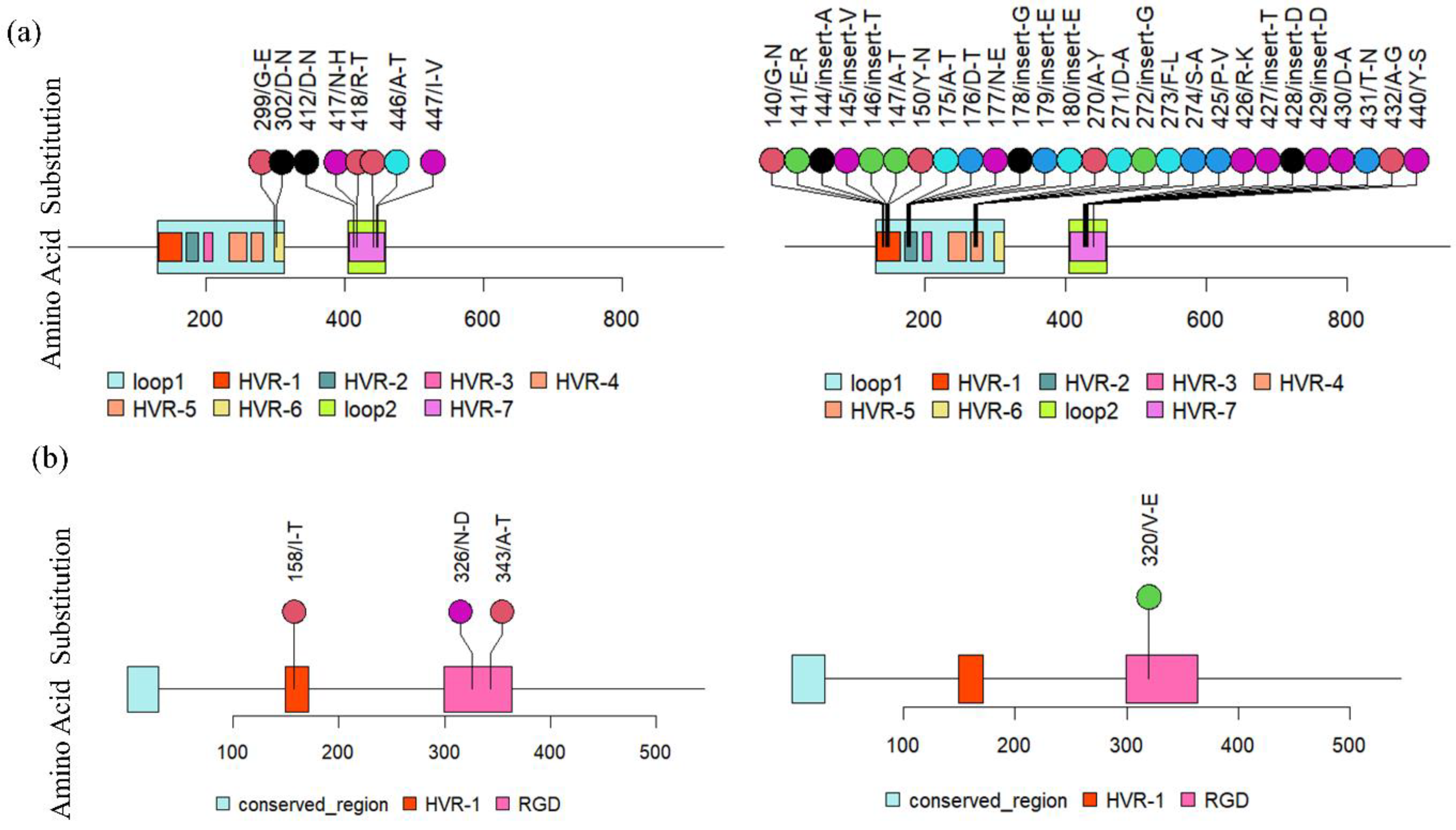
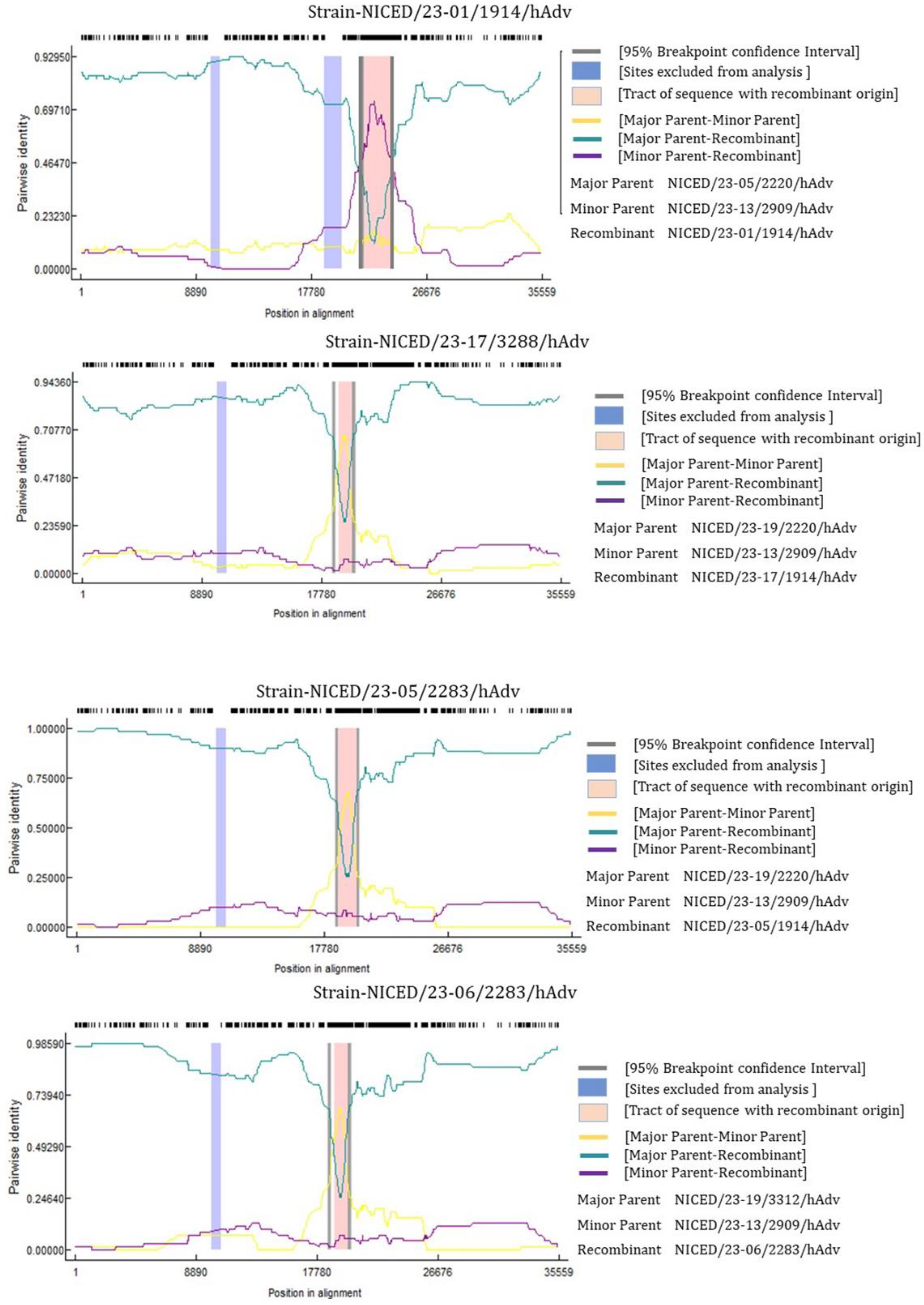
3.4. Recombinant Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crenshaw, B.J.; Jones, L.B.; Bell, C.R.; Kumar, S.; Matthews, Q.L. Perspective on Adenoviruses: Epidemiology, Pathogenicity, and Gene Therapy. Biomedicines 2019, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Nishikawaji, Y.; Kawakami, H.; Kosai, K.I. Adenovirus Biology, Recombinant Adenovirus, and Adenovirus Usage in Gene Therapy. Viruses 2021, 13, 2502. [Google Scholar] [CrossRef] [PubMed]
- Benko, M.; Aoki, K.; Arnberg, N.; Davison, A.J.; Echavarria, M.; Hess, M.; Jones, M.S.; Kajan, G.L.; Kajon, A.E.; Mittal, S.K.; et al. ICTV Virus Taxonomy Profile: Adenoviridae 2022. J. Gen. Virol. 2022, 103, 001721. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Lu, R.; Zhao, Y.; Xie, Z.; Shen, J.; Tan, W. Phylogenetic Evidence for Intratypic Recombinant Events in a Novel Human Adenovirus C That Causes Severe Acute Respiratory Infection in Children. Sci. Rep. 2016, 6, 23014. [Google Scholar] [CrossRef] [PubMed]
- Kumthip, K.; Khamrin, P.; Ushijima, H.; Maneekarn, N. Enteric and Non-Enteric Adenoviruses Associated with Acute Gastroenteritis in Pediatric Patients in Thailand, 2011 to 2017. PLoS ONE 2019, 14, e0220263. [Google Scholar] [CrossRef] [PubMed]
- Proenca-Modena, J.L.; de Souza Cardoso, R.; Criado, M.F.; Milanez, G.P.; de Souza, W.M.; Parise, P.L.; Bertol, J.W.; de Jesus, B.L.S.; Prates, M.C.M.; Silva, M.L.; et al. Human Adenovirus Replication and Persistence in Hypertrophic Adenoids and Palatine Tonsils in Children. J. Med. Virol. 2019, 91, 1250–1262. [Google Scholar] [CrossRef] [PubMed]
- Mennechet, F.J.D.; Paris, O.; Ouoba, A.R.; Salazar Arenas, S.; Sirima, S.B.; Takoudjou Dzomo, G.R.; Diarra, A.; Traore, I.T.; Kania, D.; Eichholz, K.; et al. A Review of 65 Years of Human Adenovirus Seroprevalence. Expert Rev. Vaccines 2019, 18, 597–613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, W.; Zhou, X.; Zhao, Q.; Wang, Q.; Jia, B. Seroprevalence of Neutralizing Antibodies to Human Adenoviruses Type-5 and Type-26 and Chimpanzee Adenovirus Type-68 in Healthy Chinese Adults. J. Med. Virol. 2013, 85, 1077–1084. [Google Scholar] [CrossRef]
- Akello, J.O.; Kamgang, R.; Barbani, M.T.; Suter-Riniker, F.; Aebi, C.; Beuret, C.; Paris, D.H.; Leib, S.L.; Ramette, A. Genomic Analyses of Human Adenoviruses Unravel Novel Recombinant Genotypes Associated with Severe Infections in Pediatric Patients. Sci. Rep. 2021, 11, 24038. [Google Scholar] [CrossRef]
- Adenovirus Outbreaks|CDC. Available online: https://www.cdc.gov/adenovirus/outbreaks.html (accessed on 9 June 2023).
- Jin, X.; Ren, J.; Li, R.; Gao, Y.; Zhang, H.; Li, J.; Zhang, J.; Wang, X.; Wang, G. Global Burden of Upper Respiratory Infections in 204 Countries and Territories, from 1990 to 2019. eClinicalMedicine 2021, 37, 100986. [Google Scholar] [CrossRef]
- Majumdar, A.; Saha, R.; Chatterjee, A.; Gupta, R.; Chaudhuri, R.D.; Chakrabarti, A.K.; Chawla-Sarkar, M.; Dutta, S. Upsurge in Hospitalization of Pediatric Patients with Severe Acute Respiratory Infections in Kolkata and Surrounding Districts Caused by Recombinant Human Respiratory Adenovirus Type B 7/3. J. Med. Virol. 2023, 95, e28897. [Google Scholar] [CrossRef]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.; Korobeynikov, A.; Lapidus, A.; Prjibelsky, A.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling Genomes and Mini-Metagenomes from Highly Chimeric Reads. In Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer: Berlin/Heidelberg, Germany, 2013; Volume 7821, pp. 158–170. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Feng, C.; Han, R.; Wang, Z.; Ye, L.; Du, Z.; Wei, H.; Zhang, F.; Peng, Z.; Yang, J. TrRosettaRNA: Automated Prediction of RNA 3D Structure with Transformer Network. Nat. Commun. 2023, 14, 7266. [Google Scholar] [CrossRef] [PubMed]
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (Force-Directed RNA): Simple and Effective Online RNA Secondary Structure Diagrams. Bioinformatics 2015, 31, 3377. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. HDOCK: A Web Server for Protein-Protein and Protein-DNA/RNA Docking Based on a Hybrid Strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]
- Yan, Y.; Wen, Z.; Wang, X.; Huang, S.Y. Addressing Recent Docking Challenges: A Hybrid Strategy to Integrate Template-Based and Free Protein-Protein Docking. Proteins 2017, 85, 497–512. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The HDOCK Server for Integrated Protein–Protein Docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zou, X. An Iterative Knowledge-Based Scoring Function for Protein-Protein Recognition. Proteins 2008, 72, 557–579. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zou, X. A Knowledge-Based Scoring Function for Protein-RNA Interactions Derived from a Statistical Mechanics-Based Iterative Method. Nucleic Acids Res. 2014, 42, e55. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, J.; Fraile, A.; Sacristán, S.; Malpica, J.M.; García-Arenal, F. Role of Recombination in the Evolution of Natural Populations of Cucumber Mosaic Virus, a Tripartite RNA Plant Virus. Virology 2005, 332, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, A.; Hage, E.; Ganzenmueller, T.; Böttcher, S.; Hofmann, J.; Hamprecht, K.; Obermeier, P.; Rath, B.; Hausmann, F.; Dobner, T.; et al. Molecular Evolution of Human Adenovirus (HAdV) Species C. Sci. Rep. 2019, 9, 1039. [Google Scholar] [CrossRef]
- Walsh, M.P.; Chintakuntlawar, A.; Robinson, C.M.; Madisch, I.; Harrach, B.; Hudson, N.R.; Schnurr, D.; Heim, A.; Chodosh, J.; Seto, D.; et al. Evidence of Molecular Evolution Driven by Recombination Events Influencing Tropism in a Novel Human Adenovirus That Causes Epidemic Keratoconjunctivitis. PLoS ONE 2009, 4, e5635. [Google Scholar] [CrossRef]
- Crawford-Miksza, L.K.; Schnurr, D.P. Adenovirus Serotype Evolution Is Driven by Illegitimate Recombination in the Hypervariable Regions of the Hexon Protein. Virology 1996, 224, 357–367. [Google Scholar] [CrossRef]
- Ebner, K.; Pinsker, W.; Lion, T. Comparative Sequence Analysis of the Hexon Gene in the Entire Spectrum of Human Adenovirus Serotypes: Phylogenetic, Taxonomic, and Clinical Implications. J. Virol. 2005, 79, 12635–12642. [Google Scholar] [CrossRef]
- Duan, Y.; Xu, B.; Li, C.; Bao, Y.; An, S.; Zhou, Y.; Chen, A.; Deng, L.; Ning, L.; Zhu, Y.; et al. Molecular Characteristics of Human Adenovirus Type 3 Circulating in Parts of China During 2014–2018. Front. Microbiol. 2021, 12, 688661. [Google Scholar] [CrossRef]
- Xu, W.; Erdman, D.D. Type-Specific Identification of Human Adenovirus 3, 7, and 21 by a Multiplex PCR Assay. J. Med. Virol. 2001, 64, 537–542. [Google Scholar] [CrossRef]
- Wickham, T.J.; Filardo, E.J.; Cheresh, D.A.; Nemerow, G.R. Integrin Alpha v Beta 5 Selectively Promotes Adenovirus Mediated Cell Membrane Permeabilization. J. Cell Biol. 1994, 127, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Hood, I.V.; Gordon, J.M.; Bou-Nader, C.; Henderson, F.E.; Bahmanjah, S.; Zhang, J. Crystal Structure of an Adenovirus Virus-Associated RNA. Nat. Commun. 2019, 10, 2871. [Google Scholar] [CrossRef] [PubMed]
- Launer-Felty, K.; Cole, J.L. Domain Interactions in Adenovirus VAI RNA Mediate High-Affinity PKR Binding. J. Mol. Biol. 2014, 426, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, C.; Peng, Z.; Zhao, J.; Gong, G.; Tan, D. MiR-197 Expression in Peripheral Blood Mononuclear Cells from Hepatitis B Virus-Infected Patients. Gut Liver 2013, 7, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Kaplanski, G. Interleukin-18: Biological Properties and Role in Disease Pathogenesis. Immunol. Rev. 2018, 281, 138–153. [Google Scholar] [CrossRef]
- Oliveira, E.R.A.; Bouvier, M. Immune Evasion by Adenoviruses: A Window into Host–Virus Adaptation. FEBS Lett. 2019, 593, 3496–3503. [Google Scholar] [CrossRef]
- Sester, M.; Ruszics, Z.; Mackley, E.; Burgert, H.-G. The Transmembrane Domain of the Adenovirus E3/19K Protein Acts as an Endoplasmic Reticulum Retention Signal and Contributes to Intracellular Sequestration of Major Histocompatibility Complex Class I Molecules. J. Virol. 2013, 87, 6104–6117. [Google Scholar] [CrossRef] [PubMed]
- Arnberg, N. Adenovirus E3 Protein Modulates Leukocyte Functions. Proc. Natl. Acad. Sci. USA 2013, 110, 19976–19977. [Google Scholar] [CrossRef]
- Mautner, V.; Mackay, N. Recombination in Adenovirus: Analysis of Crossover Sites in Intertypic Overlap Recombinants. Virology 1984, 139, 43–52. [Google Scholar] [CrossRef]
- Williams, J.; Grodzicker, T.; Sharp, P.; Sambrook, J. Adenovirus Recombination: Physical Mapping of Crossover Events. Cell 1975, 4, 113–119. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. | Patient | WG Homology | Genotype Based on, Hexon Fiber, Penton Genes | Co-Infection | ICU | Oxygen | Final |
|---|---|---|---|---|---|---|---|
| No | ID | Admission | Requirement | Outcome | |||
| 1 | 1914 | 3 (KF268210.1, 99.21%) | 3[H3F3P7] | No | No | No | Recovered |
| 2 | 1908 | 7 (KF268125.1, 99.25%) | 7[H7F3P7] | No | Yes | Yes | Recovered |
| 3 | 2280 | 7 (KF268125.1, 99.21%) | 7[H7F3P7] | PIV | Yes | Yes | Death |
| 4 | 2283 | 7 (KF268125.1, 99.25%) | 7[H7F3P7] | No | Yes | Yes | Death |
| 5 | 2220 | 7 (KF268125.1, 99.14%) | 7[H7F3P7] | No | Yes | Yes | Death |
| 6 | 2213 | 7 (KF268125.1, 99.28%) | 7[H7F3P7] | Rhino | No | No | Recovered |
| 7 | 1995 | 7 (KF268125.1, 99.26%) | 7[H7F3P7] | No | No | No | Recovered |
| 8 | 1836 | 7 (KF268125.1, 99.16%) | 7[H7F3P7] | No | No | Yes | Recovered |
| 9 | 1639 | 7 (KF268125.1, 99.24%) | 7[H7F3P7] | No | Not Available | Not Available | Not Available |
| 10 | 2640 | 7 (KF268125.1, 99.19%) | 7[H7F3P7] | No | Yes | Yes | Death |
| 11 | 2788 | 7 (KF268125.1, 99.23%) | 7[H7F3P7] | No | Not Available | Not Available | Not Available |
| 12 | 2908 | 3 (KF268132, 97.75%) | 3[H7F3P7] | No | Yes | Yes | Recovered |
| 13 | 2909 | 3 (AY599834.1, 99.35%) | 3[H3F3P3] | No | Yes | Yes | Recovered |
| 14 | 2928 | 7 (KF268125.1, 99.19%) | 7[H7F3P7] | No | Not Available | Not Available | Not Available |
| 15 | 2986 | 7 (KF268125.1, 99.22%) | 7[H7F3P7] | PIV | Yes | Yes | Recovered |
| 16 | 3234 | 7 (KF268125.1, 99.16%) | 7[H7F3P7] | No | Yes | Yes | Death |
| 17 | 3288 | 7 (KF268125.1, 99.17%) | 7[H7F3P7] | No | No | No | Recovered |
| 18 | 3289 | 7 (KF268125.1, 99.10%) | 7[H7F3P7] | RSV | Yes | Yes | Death |
| 19 | 3312 | 7 (KF268125.1, 98.84%) | 7[H3F3P7] | No | Yes | Yes | Death |
| 20 | 3071 | 7 (KF268125.1, 99.21%) | 7[H7F3P7] | No | No | No | Recovered |
| 21 | 3073 | 7 (KF268125.1, 99.18%) | 7[H7F3P7] | No | Not Available | Not Available | Not Available |
| 22 | 2981 | 7 (KF268125.1, 99.14%) | 7[H7F3P7] | No | No | No | Recovered |
| 23 | 130 | 7 (KF268125.1, 99.05%) | 7[H7F3P7] | No | Yes | Yes | Recovered |
| 24 | 151 | 7 (KF268125.1, 99.21%) | 7[H7F3P7] | No | No | No | Recovered |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatterjee, A.; Bhattacharjee, U.; Gupta, R.; Debnath, A.; Majumdar, A.; Saha, R.; Chawla-Sarkar, M.; Chakrabarti, A.K.; Dutta, S. Genomic Expedition: Deciphering Human Adenovirus Strains from the 2023 Outbreak in West Bengal, India: Insights into Viral Evolution and Molecular Epidemiology. Viruses 2024, 16, 159. https://doi.org/10.3390/v16010159
Chatterjee A, Bhattacharjee U, Gupta R, Debnath A, Majumdar A, Saha R, Chawla-Sarkar M, Chakrabarti AK, Dutta S. Genomic Expedition: Deciphering Human Adenovirus Strains from the 2023 Outbreak in West Bengal, India: Insights into Viral Evolution and Molecular Epidemiology. Viruses. 2024; 16(1):159. https://doi.org/10.3390/v16010159
Chicago/Turabian StyleChatterjee, Ananya, Uttaran Bhattacharjee, Rudrak Gupta, Ashis Debnath, Agniva Majumdar, Ritubrita Saha, Mamta Chawla-Sarkar, Alok Kumar Chakrabarti, and Shanta Dutta. 2024. "Genomic Expedition: Deciphering Human Adenovirus Strains from the 2023 Outbreak in West Bengal, India: Insights into Viral Evolution and Molecular Epidemiology" Viruses 16, no. 1: 159. https://doi.org/10.3390/v16010159