
Herpesviridae, Neurodegenerative Disorders and Autoimmune Diseases: What Is the Relationship between Them?


Abstract
:1. Introduction
2. Neurodegenerative Diseases
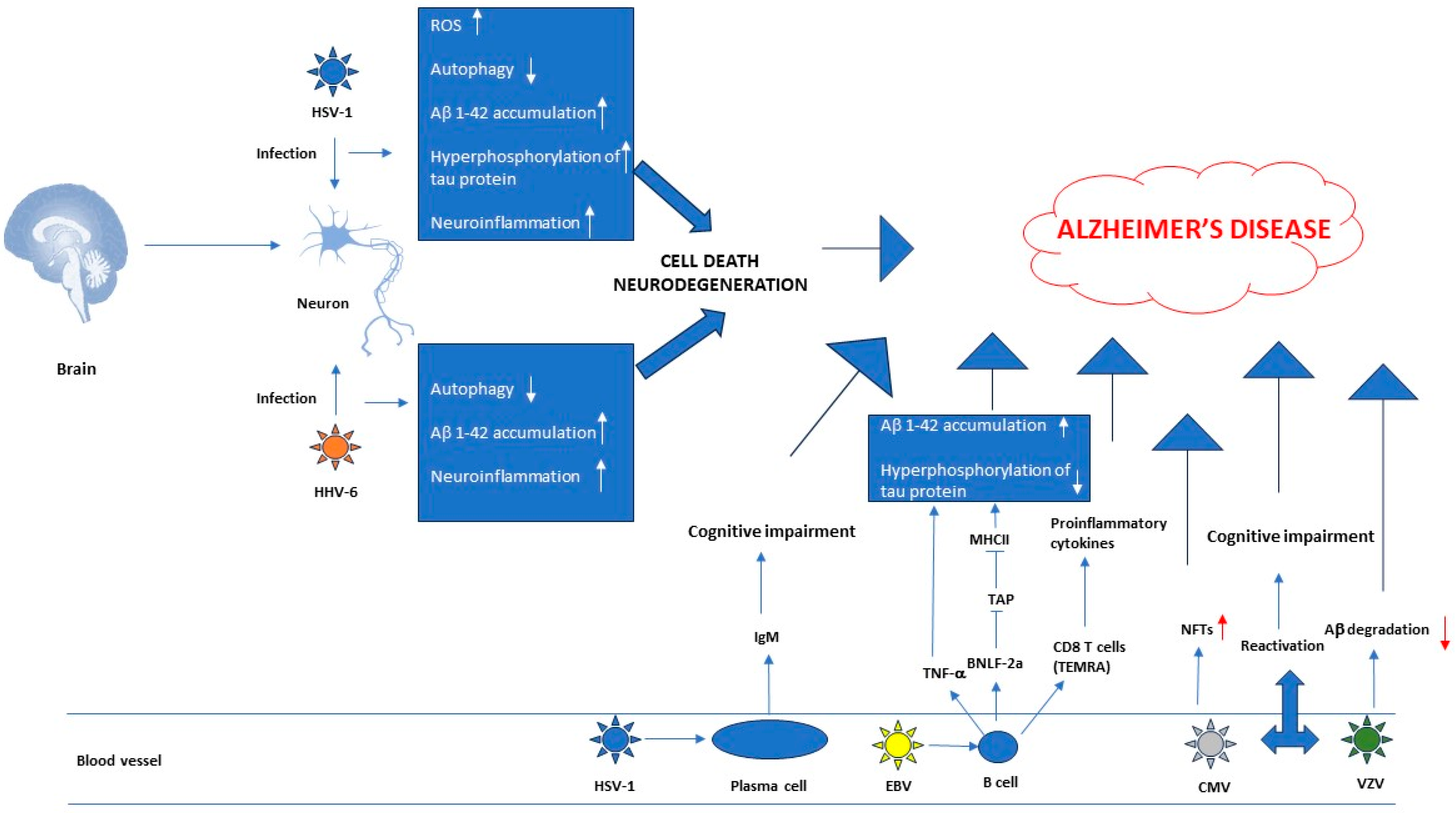
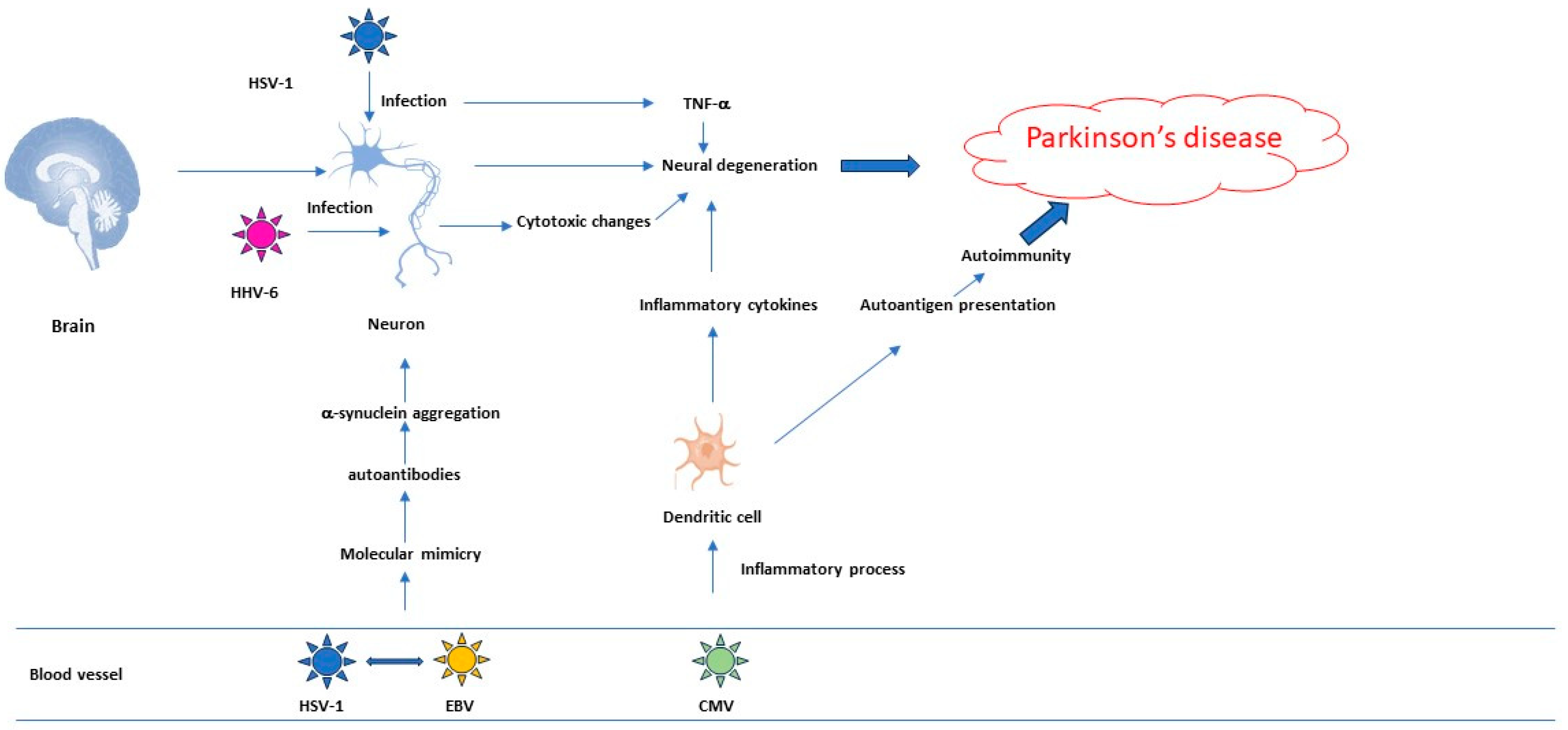
2.1. Herpesviruses’ Role in the Pathogenesis of Neurodegenerative Diseases
2.2. Research Data That Establish a Link between AD and Herpesviruses
2.3. Research Data That Establish a Link between PD and Herpesviruses
3. Autoimmune Diseases
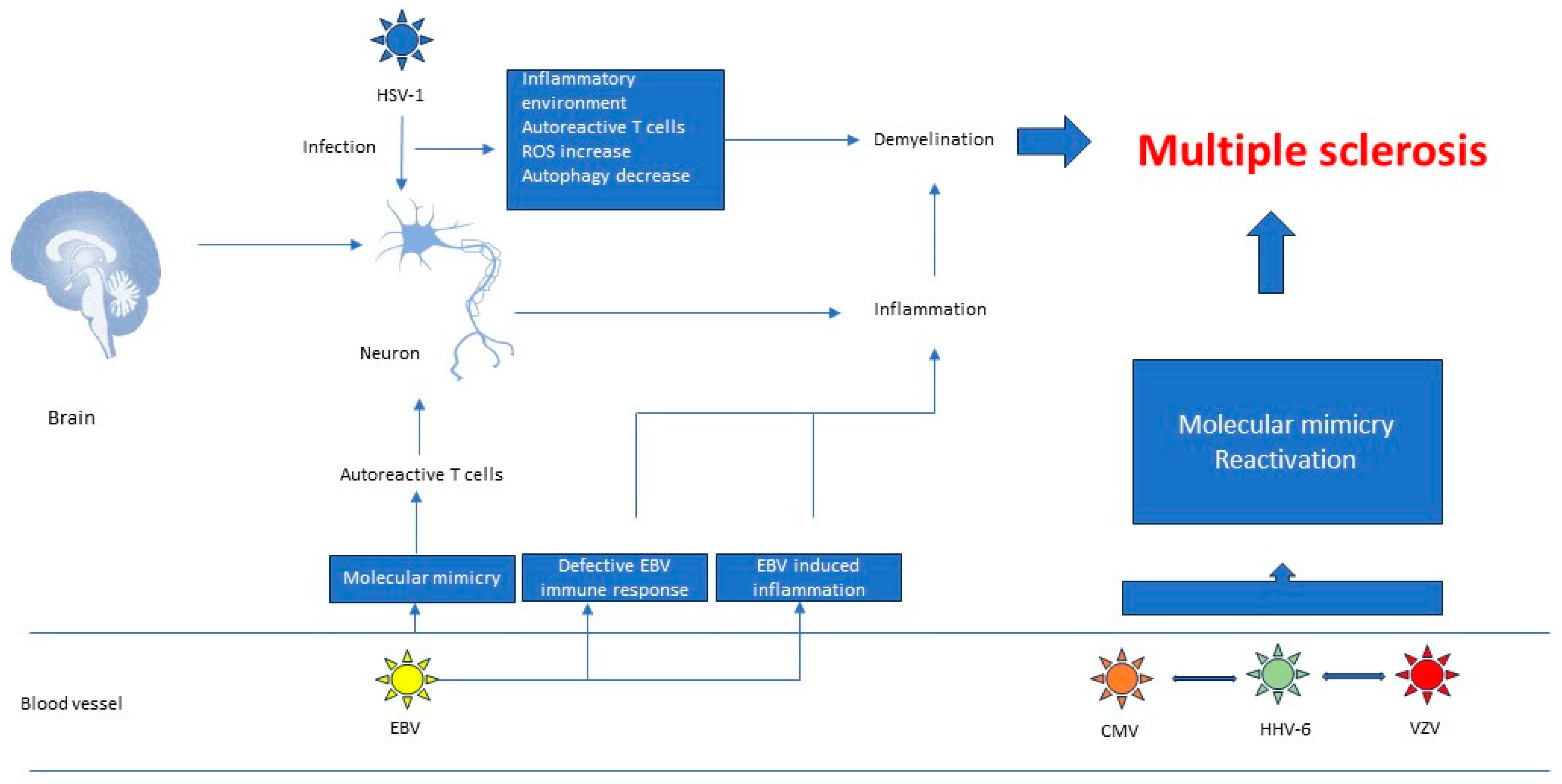
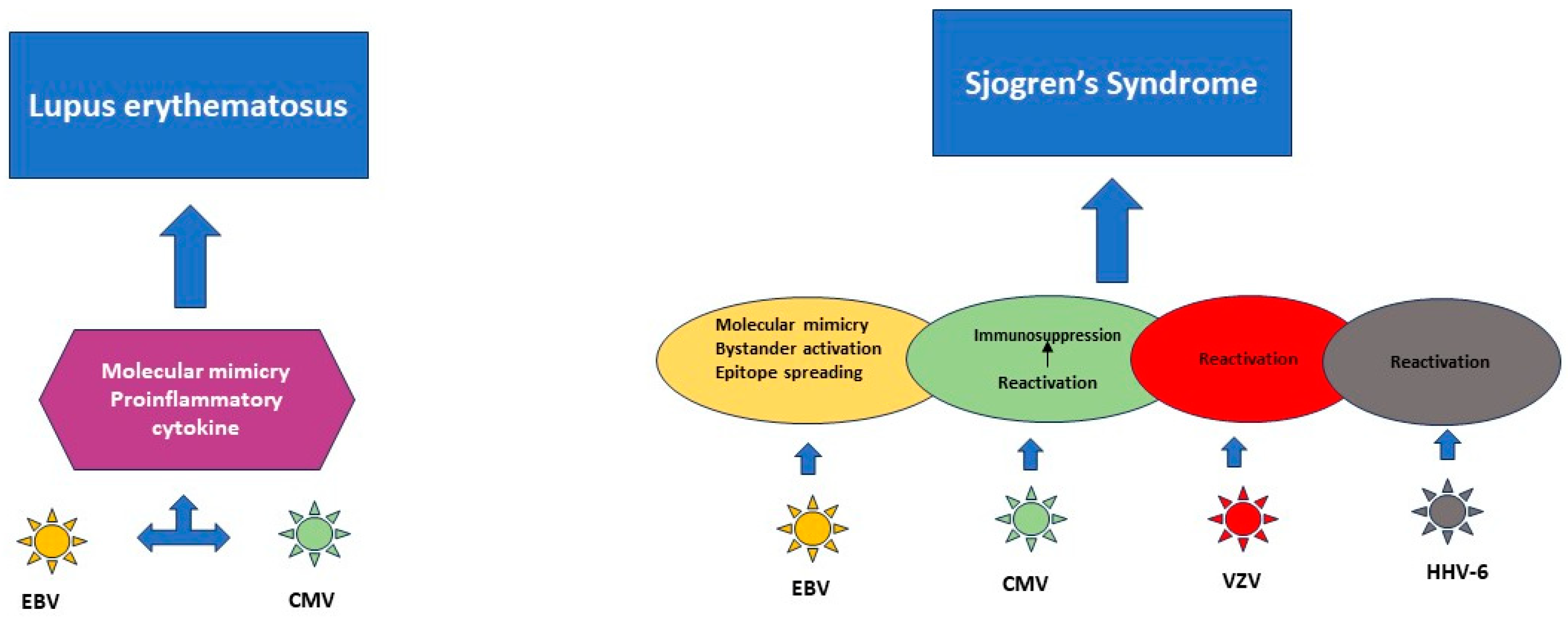
3.1. Herpesviruses’ Role in the Pathogenesis of Autoimmune Diseases
3.2. Research Data That Establish a Link between Multiple Sclerosis and Herpesviruses
3.2.1. Molecular Mimicry
3.2.2. Defective EBV Immune Responses
3.2.3. EBV Induced Inflammation
3.3. Research Data That Establish a Link between Systemic Lupus Erythematosus and Herpesviruses
3.4. Research Data That Establish a Link between Sjögren’s Syndrome and Herpesviruses
4. Relationship between Herpesviruses, Neurological Disorders and Autoimmune Diseases
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lan, K.; Luo, M.H. Herpesviruses: Epidemiology, pathogenesis, and interventions. Virol. Sin. 2017, 32, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Nath, A. Herpes viruses, Alzheimer’s disease, and related dementias: Unifying or confusing hypothesis? Neurotherapeutics 2019, 16, 180–181. [Google Scholar] [CrossRef] [PubMed]
- Rossi, B.; Santos-Lima, B.; Terrabuio, E.; Zenaro, E.; Constantin, G. Common Peripheral Immunity Mechanisms in Multiple Sclerosis and Alzheimer’s Disease. Front. Immunol. 2021, 12, 639369. [Google Scholar] [CrossRef]
- Bulté, D.; Rigamonti, C.; Romano, A.; Mortellaro, A. Inflammasomes: Mechanisms of Action and Involvement in Human Diseases. Cells 2023, 12, 1766. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, J.J.; Pröbstel, A.K.; Zamvil, S.S. B cells in autoimmune and neurodegenerative central nervous system disease. Nat. Rev. Neurosci. 2019, 20, 728–745. [Google Scholar] [CrossRef]
- Chen, H.; Ritz, B. The search for environmental causes of Parkinson’s disease: Moving forward. J. Parkinons Dis. 2018, 8, S9–S17. [Google Scholar] [CrossRef]
- Marras, C.; Canning, C.G.; Goldman, S.M. Environment, lifestyle, and Parkinson’s disease: Implications for prevention in the next decade. Mov. Disord. 2019, 34, 801–811. [Google Scholar] [CrossRef]
- Patrick, K.L.; Bell, S.L.; Weindel, C.G.; Watson, R.O. Exploring the “Multiple-hit hypothesis” of neurodegenerative disease: Bacterial infection comes up to bat. Front. Cell. Infect. Microbiol. 2019, 9, 138. [Google Scholar] [CrossRef]
- Wouk, J.; Zendrini Rechenchonski, D.; Cerqueira Dias Rodrigues, B.; Vicente Ribelato, E.; Faccin-Galhardi, L.C. Viral infections and their relationship to neurological disorders. Arch. Virol. 2021, 166, 733–753. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Clavaguera, F.; Potier, M.C. The prion-like propagation hypothesis in Alzheimer’s and Parkinson’s disease. Curr. Opin. Neurol. 2019, 32, 266–271. [Google Scholar] [CrossRef]
- Shi, M.; Li, C.; Tain, X.; Chu, F.; Zhu, J. Can control infections slow down the progression of Alzheimer’s disease? Talking about the role of infections in Alzheimer’s disease. Front. Aging Neurosci. 2021, 13, 434. [Google Scholar] [CrossRef]
- Patterson, C. World Alzheimer Report 2018; Alzheimer’s Disease International: London, UK, 2018. [Google Scholar]
- Parkinsons’s Foundation. Available online: https://www.parkinson.org/understandingParkinsons/Statistics (accessed on 2 August 2020).
- de Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. Biomarkers for Alzheimer’s disease: Current status and prospects for the future. J. Intern. Med. 2018, 284, 643–663. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, T.; Bu, G.; Xu, H. Dysregulation of protein trafficking in neurodegeneration. Mol. Neurodegener. 2014, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, G.; Dolciotti, C.; Picchi, L.; Bonuccelli, U. Alzheimer and his disease: A brief history. Neurol. Sci. 2011, 32, 275–279. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Kolarova, M.; Garcia-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Sun, C. Calcium and neurogenesis in Alzheimer’s disease. Front. Neurosci. 2010, 4, 194. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 8, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Welikovitch, L.A.; Do Carmo, S.; Magloczky, Z.; Malcolm, J.C.; Loke, J.; Klein, W.L.; Freund, T.; Cuello, A.C. Early intraneuronal amyloid triggers neuron derived inflammatory signaling in APP transgenic rats and human brain. Proc. Natl. Acad. Sci. USA 2020, 117, 6844–6854. [Google Scholar] [CrossRef]
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, lifestyle stress, and neurodegeneration. Immunity 2020, 52, 222–240. [Google Scholar] [CrossRef]
- Laval, K.; Enquist, L.W. The potential role of herpes simplex virus type 1 and neuroinflammation in the pathogenesis of Alzheimer’s disease. Front. Neurol. 2021, 12, 458. [Google Scholar] [CrossRef]
- Grosch, J.; Winkler, J.; Kohl, Z. Early degeneration of both dopaminergic and serotnergic axons—A common mechanism in Parkinson’s disease. Front. Cell Neurosci. 2016, 10, 293. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Steur, E.N.H.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.; Ho, P.W.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The Interplay of Aging, Genetics and Environmental Factors in the Pathogenesis of Parkinson’s Disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Kent, K.; Pierce, A.; Leung, C.; Kang, M.S.; Okada, H.; Honig, L.; Vonsattel, J.P.; Kim, T.W. Model-Guided MicroarrayImplicates the Retromer Complex in Alzheimer’s Disease. Ann. Neurol. 2005, 58, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Tang, F.L.; Hong, Y.; Luo, S.W.; Wang, C.L.; He, W.; Shen, C.; Jung, J.U.; Xiong, F.; Lee, D.H.; et al. Vps35 HaploinsufficiencyIncreases Alzheimer’s Disease Neuropathology. J. Cell Biol. 2011, 195, 765–779. [Google Scholar] [CrossRef]
- Li, J.G.; Chiu, J.; Pratico, D. Full Recovery of the Alzheimer’s Disease Phenotype by Gain of Function of Vacuolar Protein Sorting 35. Mol. Psychiatry 2020, 25, 2630–2640. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.E.; Healy, M.D.; Collins, B.M. Towards a Molecular Understanding of Endosomal Trafficking by Retromer and Retriever. Traffic 2019, 20, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Cullen, P.J.; Steinberg, F. To Degrade or Not to Degrade: Mechanisms and Significance of Endocytic Recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Temkin, P.; Lauffer, B.; Jager, S.; Cimermancic, P.; Krogan, N.J.; von Zastrow, M. Snx27 Mediates Retromer Tubule Entry and Endosome-to-Plasma Membrane Trafficking of Signalling Receptors. Nat. Cell Biol. 2011, 13, 715–721. [Google Scholar] [CrossRef]
- Tang, F.L.; Erion, J.R.; Tian, Y.; Liu, W.; Yin, D.M.; Ye, J.; Tang, B.; Mei, L.; Xiong, W.C. for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for Alpha-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. 2015, 35, 10613–10628. [Google Scholar] [CrossRef]
- Cui, Y.; Yang, Z.; Flores-Rodriguez, N.; Follett, J.; Ariotti, N.; Wall, A.A.; Parton, R.G.; Teasdale, R.D. Formation of Retromer Transport Carriers Is Disrupted by the Parkinson Disease-Linked Vps35 D620n Variant. Traffic 2021, 22, 123–136. [Google Scholar] [CrossRef]
- Dando, S.J.; Mackay-Sim, A.; Norton, R.; Currie, B.J.; St John, J.A.; Ekberg, J.A.K.; Batzloff, M.; Ulett, G.C.; Beacham, I.R. Pathogens penetrating the central nervous system: Infection pathways and the cellular and molecular mechanisms of invasion. Clin. Microbiol. Rev. 2014, 27, 691–726. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S. Mechanisms of microbial traversal of the blood-brain barrier. Nat. Rev. Microbiol. 2008, 6, 625–634. [Google Scholar] [CrossRef]
- D’ Mello, C.; Swain, M.G. Immune-to-brain communication pathways in inflammation-associated sickness and depression. In Current Topics in Behavioral Neurosciences; Dantzer, R., Capuron, L., Eds.; Springer: Cham, Switzerland, 2017; pp. 73–94. [Google Scholar]
- McGavern, D.B.; Kang, S.S. Illuminating viral infections in the nervous system. Nat. Rev. Immunol. 2011, 11, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Van Hoecke, L.; Vandenbroucke, R. The impact of systemic inflammation on Alzheimer’s disease pathology. Front. Immunol. 2021, 12, 79867. [Google Scholar] [CrossRef] [PubMed]
- Kirkley, K.S.; Popichak, K.A.; Afzali, M.F.; Legare, M.E.; Tjalkens, R.B. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J. Neuroinflamm. 2017, 14, 99. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Orellana, D.I.; González-Billault, C.; Maccioni, R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef]
- Woulfe, J.M.; Gray, M.T.; Gray, D.A.; Munoz, D.G.; Middeldorp, J.M. Hypothesis: A role for EBV-induced molecular mimicry in Parkinson’s disease. Parkinsonism Relat. Disord. 2014, 20, 685–694. [Google Scholar] [CrossRef]
- De Chiara, G.; Piacentini, R.; Fabiani, M.; Mastrodonato, A.; Marcocci, M.E.; Limingi, D.; Napoletani, G.; Protto, V.; Coluccio, P.; Celestino, I.; et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 2019, 15, e1007617. [Google Scholar] [CrossRef]
- Ezzat, K.; Pernemalm, M.; Palsson, S.; Roberts, T.C.; Jarver, P.; Dondalska, A.; Bestas, B.; Sobkowiak, M.J.; Levanen, B.; Skold, M.; et al. The viral protein corona directs viral pathogenesis and amyloid aggregation. Nat. Commun. 2019, 10, 2331. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Frost, A.L.; Itzaki, R.F. Alzheimer’s disease-specific tau phosphorylation is induced by herpes simplex virus type 1. J. Alzheimers Dis. 2009, 16, 341–350. [Google Scholar] [CrossRef]
- Itzhaki, R.F.; Golde, T.E.; Heneka, M.T.; Readhead, B. Do infections have a role in the pathogenesis of Alzheimer disease? Nat. Rev. Neurol. 2020, 1, 193–197. [Google Scholar] [CrossRef]
- Jamieson, G.A.; Maitland, N.J.; Craske, J.; Wilcock, G.K.; Itzhaki, R.F. Detection of herpes simplex virus type 1 DNA sequences in normal and Alzheimer’s disease brain using polymerase chain reaction. Biochem. Soc. Trans. 1991, 19, 122S. [Google Scholar] [CrossRef]
- Itzhaki, R.F.; Lin, W.R.; Shang, D.; Wilcock, G.K.; Faragher, B.; Jamieson, G.A. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 1997, 349, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Mee, A.P.; Itzhaki, R.F. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 2009, 217, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Zare-Shahabadi, A.; Masliah, E.; Johnson, G.V.W.; Rezaei, N. Autophagy in Alzheimer’s disease. Rev. Neurosci. 2015, 26, 385–395. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Degradation of tau protein by autophagy and proteasomal pathways. Biochem. Soc. Trans. 2012, 40, 644–652. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s disease: Cellular and molecular mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s disease: From molecular mechanisms to therapeutic implications. Front. Aging Neurosci. 2018, 10, 04. [Google Scholar] [CrossRef]
- Hill, S.M.; Wrobel, L.; Rubinsztein, D.C. Post-translational modifications of Beclin 1 provide multiple strategies for autophagy regulation. Cell Death Differ. 2019, 26, 617–629. [Google Scholar] [CrossRef]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Lussignol, M.; Queval, C.; Bernet-Camard, M.F.; Cotte-Laffitte, J.; Beau, I.; Codogno, P.; Esclatine, A. The herpes simplex virus1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J. Virol. 2013, 87, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Kavouras, J.H.; Prandovszky, E.; Valyi-Nagy, K.; Kovacs, S.K.; Tiwari, V.; Kovacs, M.; Shukla, D.; Valyi-Nagy, T. Herpes simplex virus type 1 infection induces oxidative stress and the release of bioactive lipid peroxidation by-products in mouse P19N neural cell cultures. J. Neurovirol. 2007, 13, 416–425. [Google Scholar] [CrossRef]
- Santana, S.; Sastre, I.; Recuero, M.; Bullido, M.J.; Aldudo, J. Oxidative stress enhances neurodegeneration markers induced by herpes simplex virus type 1 infection in human neuroblastoma cells. PLoS ONE 2013, 8, e75842. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genom. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef]
- Kuhlmann, I.; Minihane, A.M.; Huebbe, P.; Nebel, A.; Rimbach, G. Apolipoprotein E genotype and hepatitis C, HIV and herpes simplex disease risk: A literature review. Lipids Health Dis. 2010, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Lovheim, H.; Norman, T.; Weidung, B.; Olsson, J.; Josefsson, M.; Adolfsson, R.; Nyberg, L.; Elgh, F. Herpes simplex virus, APOE epsilon4, and cognitive decline in old age: Results from the Betula Cohort Study. J. Alzheimers Dis. 2019, 67, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.B.; Ferland, P.; Webster, P.; Bearer, E.L. Herpes simplex virus dances with amyloid precursor protein while exiting the cell. PLoS ONE 2011, 6, e17966. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, E. Amyloid-Beta1-42 cross-reactive antibody prevalent in human sera may contribute to intraneuronal deposition of A-Beta-P-42. Int. J. Alzheimers Dis. 2018, 2018, 1672568. [Google Scholar]
- Letenneur, L.; Peres, K.; Fleury, H.; Garrigue, I.; Barberger-Gateau, P.; Helmer, C.; Orgogozo, J.M.; Gauthier, S.; Dartigues, J.F. Seropositivity to herpes simplex virus antibodies and risk of Alzheimer’s disease: A population-based cohort study. PLoS ONE 2008, 3, e3637. [Google Scholar] [CrossRef]
- Tzeng, N.S.; Chung, C.H.; Lin, F.H.; Chiang, C.P.; Yeh, C.B.; Huang, S.Y.; Lu, R.B.; Chang, H.A.; Kao, Y.C.; Yeh, H.W.; et al. Anti-herpetic medications and reduced risk of dementia in patients with herpes simplex virus infections—A nationwide, population-based cohort study in Taiwan. Neurotherapeutics 2018, 15, 417–429. [Google Scholar] [CrossRef]
- Carbone, I.; Lazzarotto, T.; Ianni, M.; Porcellini, E.; Forti, P.; Masliah, E.; Gabrielli, L.; Licastro, F. Herpes virus in Alzheimer’s disease: Relation to progression of the disease. Neurobiol. Aging 2014, 35, 122–129. [Google Scholar] [CrossRef]
- Shim, S.M.; Cheon, H.S.; Jo, C.; Koh, Y.H.; Song, J.; Jeon, J.P. Elevated Epstein-Barr Virus Antibody Level is Associated with Cognitive Decline in the Korean Elderly. J. Alzheimers Dis. 2017, 55, 293–301. [Google Scholar] [CrossRef]
- Dezfulian, M. A new Alzheimer’s disease cell model using B cells to induce beta amyloid plaque formation and increase TNF alpha expression. Int. Immunopharmacol. 2018, 59, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Ounanian, A.; Guilbert, B.; Seigneurin, J.M. Characteristics of Epstein-Barr virus transformed B cell lines from patients with Alzheimer’s disease and age-matched controls. Mech. Ageing Dev. 1992, 63, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef]
- Hur, J.Y.; Frost, G.R.; Wu, X.; Crump, C.; Pan, S.J.; Wong, E.; Barros, M.; Li, T.; Nie, P.; Zhai, Y.; et al. The innate immunity protein IFITM3 modulates gamma-secretase in Alzheimer’s disease. Nature 2020, 586, 735–740. [Google Scholar] [CrossRef]
- Tiwari, D.; Singh, V.K.; Baral, B.; Pathak, D.K.; Jayabalan, J.; Kumar, R.; Tapryal, S.; Jha, H.C. Indication of Neurodegenerative Cascade Initiation by Amyloid-like Aggregate-Forming EBV Proteins and Peptide in Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Wycisk, A.I.; Lin, J.; Loch, S.; Hobohm, K.; Funke, J.; Wieneke, R.; Koch, J.; Skach, W.R.; Mayerhofer, P.U.; Tampe, R. Epstein-Barr viral BNLF2a protein hijacks the tail-anchored protein insertion machinery to block antigen processing by the transport complex TAP. J. Biol. Chem. 2011, 286, 41402–41412. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, J.M.; Jegou, J.F.; Welsch, J.; Horvat, B. Human herpesvirus 6A infection in CD46 transgenic mice: Viral persistence in the brain and increased production of proinflammatory chemokines via TLR9. J. Virol. 2014, 88, 5421–5436. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.; Bortolotti, D.; Gentili, V.; Rotola, A.; Bolzani, S.; Caselli, E.; Tola, M.R.; Di Luca, D. KIR2DS2/KIR2DL2/HLA-C1 haplotype is associated with Alzheimer’s disease: Implication for the role of herpesvirus infections. J. Alzheimers Dis. 2019, 67, 1379–1389. [Google Scholar] [CrossRef]
- Bortolotti, D.; Gentili, V.; Rotola, A.; Caselli, E.; Rizzo, R. HHV-6 infection induces amyloid-beta expression and activation of microglial cells. Alzheimers Res. Ther. 2019, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Romeo, M.A.; Masuelli, L.; Gaeta, A.; Nazzari, C.; Granato, M.; Gilardini Montani, M.S.; Faggioni, A.; Cirone, M. Impact of HHV-6A and HHV-6B lytic infection on autophagy and endoplasmic reticulum stress. J. Gen. Virol. 2019, 100, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Romeo, M.A.; Gilardini Montani, M.S.; Gaeta, A.; D’Orazi, G.; Faggioni, A.; Cirone, M. HHV-6A infection dysregulates autophagy/UPR interplay increasing beta amyloid production and tau phosphorylation in astrocytoma cells as well as in primary neurons, possible molecular mechanisms linking viral infection to Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165647. [Google Scholar] [CrossRef] [PubMed]
- Romeo, M.A.; Gilardini Montani, M.S.; Falcinelli, L.; Gaeta, A.; Nazzari, C.; Faggioni, A.; Cirone, M. HHV-6B reduces autophagy and induces ER stress in primary monocytes impairing their survival and differentiation into dendritic cells. Virus Res. 2019, 273, 197757. [Google Scholar] [CrossRef] [PubMed]
- Costa Sa, A.C.; Madsen, H.; Brown, J.R. Shared molecular signatures across neurodegenerative diseases and herpes virus infections highlights potential mechanisms for maladaptive innate immune responses. Sci. Rep. 2019, 9, 8795. [Google Scholar] [CrossRef] [PubMed]
- Readhead, B.; Haure-Mirande, J.V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 2018, 99, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Allnutt, M.A.; Johnson, K.; Bennett, D.A.; Connor, S.M.; Troncoso, J.C.; Pletnikova, O.; Albert, M.S.; Resnick, S.M.; Scholz, S.W.; De Jager, P.L.; et al. Human herpesvirus 6 detection in Alzheimer’s disease cases and controls across multiple cohorts. Neuron 2020, 105, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.H.; Liu, Z. Are HHV-6A and HHV-7 really more abundant in Alzheimer’s disease? Neuron 2019, 104, 1034–1035. [Google Scholar] [CrossRef] [PubMed]
- Chorlton, S.D. Reanalysis of Alzheimer’s brain sequencing data reveals absence of purported HHV-6A and HHV-7. J. Bioinform. Comput. Biol. 2020, 18, 2050012. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.C.; Wu, S.I.; Huang, K.Y.; Yang, Y.H.; Kuo, T.Y.; Liang, H.Y.; Huang, K.L.; Gossop, M. Herpes zoster and dementia: A nationwide population-based cohort study. J. Clin. Psychiatry 2018, 79, 16m11312. [Google Scholar] [CrossRef]
- Tsai, M.C.; Cheng, W.L.; Sheu, J.J.; Huang, C.C.; Shia, B.C.; Kao, L.T.; Lin, H.C. Increased risk of dementia following herpes zoster ophthalmicus. PLoS ONE 2017, 12, e0188490. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ali, M.A.; Wang, K.; Sayre, D.; Hamel, F.G.; Fisher, E.R.; Bennett, R.G.; Cohen, J.I. Insulin degrading enzyme induces a con-formational change in Varicella-Zoster Virus gE, and enhances virus infectivity and stability. PLoS ONE 2010, 5, e11327. [Google Scholar]
- Bernstein, H.; Keilhoff, G.; Dobrowolny, H.; Steiner, J. Binding varicella zoster virus: An underestimated facet of insulin degrading enzyme’s implication for Alzheimer´s disease pathology? Eur. Arch. Psychiatry Clin. Neurosci. 2020, 270, 495–496. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Kwon, D.E.; Do Han, K.; La, Y.; Han, S.H. Association between cytomegalovirus end-organ diseases and moderate-to severe dementia: A population-based cohort study. BMC Neurol. 2020, 20, 216. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.E.; Haan, M.N.; Blythe, L.; Moore, K.; Gonzalez, J.M.; Jagust, W. The Influence of latent viral infection on rate of cognitive decline over 4 years. J. Am. Geriatr. Soc. 2006, 54, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Lurain, N.S.; Hanson, B.A.; Martinson, J.; Leurgans, S.E.; Landay, A.L.; Bennett, D.A.; Schneider, J.A. Virological and immunological characteristics of human cytomegalovirus infection associated with Alzheimer disease. J. Infect. Dis. 2013, 208, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Caggiu, E.; Paulus, K.; Arru, G.; Piredda, R.; Sechi, G.P.; Sechi, A. Humoral cross reactivity between α-synuclein and herpes simplex-1 epitope in Parkinson’s disease, a triggering role in the disease? J. Neuroimmunol. 2016, 291, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Caggiu, E.; Arru, G.; Hosseini, S.; Niegowska, M.; Sechi, G.; Zarbo, I.R.; Sechi, L.A. Inflammation, infectious triggers, and Parkinson’s disease. Front. Neurol. 2019, 10, 122. [Google Scholar] [CrossRef]
- Woulfe, J.; Hoogendoorn, H.; Tarnopolsky, M.; Muñoz, D.G. Monoclonal antibodies against Epstein-Barr virus cross-react with α-synuclein in human brain. Neurology 2000, 55, 1398–1440. [Google Scholar] [CrossRef] [PubMed]
- Goldeck, D.; Maetzler, W.; Berg, D.; Oettinger, L.; Pawelec, G. Altered dendritic cell subset distribution in patients with Parkinson’s disease: Impact of cmv serostatus. J. Neuroimmunol. 2016, 290, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Koutsilieri, E.; Lutz, M.B.; Scheller, C. Autoimmunity, dendritic cells and relevance for Parkinson’s disease. J. Neural Transm. 2013, 120, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Cury, R.G.; Lopez, W.O.C. Bilateral striatal lesion due to herpesvirus-6 infection. J. Neurol. Sci. 2015, 358, 538–539. [Google Scholar] [CrossRef] [PubMed]
- Dzwonek, J.; Rylski, M.; Kaczmarek, L. Matrix metalloproteinases and their endogenous inhibitors in neuronal physiology of the adult brain. FEBS Lett. 2004, 567, 129–135. [Google Scholar] [CrossRef]
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of multiple sclerosis 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Ercolini, A.M.; Miller, S.D. The role of infections in autoimmune disease. Clin. Exp. Immunol. 2009, 155, 1–15. [Google Scholar] [CrossRef]
- Temajo, N.O.; Howard, N. The mosaic of environment involvement in autoimmunity: The abrogation of viral latency by stress, a non-infectious environmental agent, is an intrinsic prerequisite prelude before viruses can rank as infectious environmental agents that trigger autoimmune diseases. Autoimmun. Rev. 2014, 13, 635–640. [Google Scholar] [PubMed]
- Dimitrov, L.G. What’ s new in multiple sclerosis. Br. J. Gen. Pract. 2014, 64, 612.e3. [Google Scholar] [CrossRef] [PubMed]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mai, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. 2020, 26, 1816–1821. [Google Scholar] [CrossRef]
- Confavreux, C.; Vukusic, S. The clinical course of multiple sclerosis. Handb. Clin. Neurol. 2014, 122, 343–369. [Google Scholar] [PubMed]
- Kular, L.; Liu, Y.; Ruhrmann, S.; Zheleznyakova, G.; Marabita, F.; Gomez-Cabrero, D.; James, T.; Ewing, E.; Lindén, M.; Górnikiewicz, B.; et al. DNA methylation as a mediator of HLA-DRB1* 15: 01 and a protective variant in multiple sclerosis. Nat. Commun. 2018, 9, 2397. [Google Scholar] [CrossRef]
- Thakolwiboon, S.; Zhao-Fleming, H.; Karukote, A.; Pachariyanon, P.; Williams, H.G.; Avila, M. Regional differences in the association of cytomegalovirus seropositivity and multiple sclerosis: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2020, 45, 102393. [Google Scholar] [CrossRef] [PubMed]
- Brodin, P.; Jojic, V.; Gao, T.; Bhattacharya, S.; Lopez Angel, C.J.; Furan, D.; Shen-Orr, S.; Dekker, C.L.; Swan, G.E.; Butte, A.J. Variation in the human immune system is largely driven by non- heritable influences. Cell 2015, 160, 37–47. [Google Scholar] [CrossRef]
- Mok, C.C.; Lau, C.S. Pathogenesis of systemic lupus erythematosus. J. Clin. Pathol. 2003, 56, 481–490. [Google Scholar] [CrossRef]
- Li, Z.X.; Zeng, S.; Wu, H.X.; Zhou, Y. The risk of systemic lupus erythematosus associated with Epstein-Barr virus infection. A systematic review and meta-analysis. Clin. Experiment. Med. 2019, 19, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Quaglia, M.; Merlotti, G.; DeAndrea, M.; Borgogna, C.; Cantaluppi, V. Viral Infections and Systemic Lupus Erythematosus: New Players in an Old Story. Viruses 2021, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Long, H.; Lu, Q. Recent Advances in Understanding Pathogenesis and Therapeutic Strategies of Systemic Lupus Erythematosus. Int. Immunopharmacol. 2020, 89, 107028. [Google Scholar] [CrossRef]
- Aringer, M.; Johnson, S.R. Classifying and Diagnosing Systemic Lupus Erythematosus in the21st Century. Rheumatology 2020, 59, v4–v11. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, H.; Yuliasih, Y. Disentangling the Pathogenesis of Systemic Lupus Erythematosus: Close Ties between Immunological, Genetic and Environmental Factors. Medicina 2023, 59, 1033. [Google Scholar] [CrossRef]
- Rigante, D.; Esposito, S. Infections and Systemic Lupus Erythematosus: Binding or Sparring Partners? Int. J. Mol. Sci. 2015, 16, 17331–17343. [Google Scholar] [CrossRef]
- Illescas-Montes, R.; Corona-Castro, C.C.; Melguizo-Rodríguez, L.; Ruiz, C.; Costela-Ruiz, V.J. Infectious Processes and Systemic Lupus Erythematosus. Immunology 2019, 158, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X.; Criswell, L.A. Primary Sjogren’s Syndrome. N. Engl. J. Med. 2018, 378, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Martin-Nares, E.; Hernandez-Molina, G. Novel autoantibodies in Sjogren’s syndrome: A comprehensive review. Autoimmun. Rev. 2019, 18, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, C.P.; Moutsopoulos, H.M. Sjogren’s syndrome. Annu. Rev. Pathol. 2014, 9, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Parisis, D.; Chivasso, C.; Perret, J.; Soyfoo, M.S.; Delporte, C. Current state of knowledge on primary sjögren’s syndrome, an autoimmune exocrinopathy. J. Clin. Med. 2020, 9, 2299. [Google Scholar] [CrossRef]
- Fisher, B.A.; Jonsson, R.; Daniels, T.; Bombardieri, M.; Brown, R.M.; Morgan, P.; Bombardieri, S.; Ng, W.F.; Tzioufas, A.G.; Vitali, C.; et al. Standardisation of labial salivary gland histopathology in clinical trials in primary Sjögren’s syndrome. Ann. Rheum. Dis. 2017, 76, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Pontarini, E.; Lucchesi, D.; Bombardieri, M. Current views on the pathogenesis of Sjögren’s syndrome. Curr. Opin. Rheumatol. 2018, 30, 215–221. [Google Scholar] [CrossRef]
- Maslinska, M.; Kostyra-Grabczak, K. The role of virus infections in Sjögren’s syndrome. Front. Immunol. 2022, 13, 823659. [Google Scholar] [CrossRef]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Molecular mimicry as a mechanism of autoimmune disease. Clin. Rev. Allerg. Immunol. 2012, 42, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Tapryal, S.; Gaur, V.; Kaur, K.J.; Salunke, D.M. Structural evaluation of a mimicry-recognizing paratope: Plasticity in antigen-antibody interactions manifests in molecular mimicry. J. Immunol. 2013, 191, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Dreyfus, D.H. Gene sharing between Epstein-Barr virus and human immune response genes. Immunol. Res. 2017, 65, 37–45. [Google Scholar] [CrossRef]
- Pacheco, Y.; Acosta-Ampudia, Y.; Monsalve, D.M.; Chang, C.; Gershwin, M.E.; Anaya, J.M. Bystander activation and autoimmunity. J. Autoimmun. 2019, 103, 102301. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.M.; Black, M.M. Epitope spreading: Protection from pathogens, but propagation of autoimmunity? Clin. Exp. Dermatol. 2001, 26, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, P.; Mancuso, R.; Pagani, E.; Guerini, F.R.; Calvo, M.G.; Saresella, M.; Speciale, L.; Caputo, D. Molecular evidences for a role of HSV-1 in multiple sclerosis clinical acute attack. J. Neurovirol. 2000, 6, S109–S114. [Google Scholar] [PubMed]
- Sanders, V.; Waddell, A.; Felisan, S.; Li, X.; Conrad, A.; Tourtellotte, W. Herpes simplex virus in postmortem multiple sclerosis brain tissue. Arch. Neurol. 1996, 53, 125–133. [Google Scholar] [CrossRef]
- Boukhvalova, M.S.; Mortensen, E.; Mbaye, A.; Lopez, D.; Kastrukoff, L.; Blanco, J.C.G. Herpes simplex virus 1 induces brain inflammation and multifocal demyelination in the cotton rat sigmodon hispidus. J. Virol. 2020, 94, 10–1128. [Google Scholar] [CrossRef]
- Waubant, E.; Mowry, E.M.; Krupp, L.; Chitnis, T.; Yeh, E.A.; Kuntz, N.; Ness, J.; Chabas, D.; Strober, J.; McDonald, J.; et al. Common viruses associated with lower pediatric multiple sclerosis risk. Neurology 2011, 76, 1989–1995. [Google Scholar] [CrossRef]
- Kastrukoff, L.F.; Lau, A.S.; Thomas, E.E. The effect of mouse strain on herpes simplex virus type 1 (HSV-1) infection of the central nervous system (CNS). Herpesviridae 2012, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Bang, D.; Lee, S.; Lee, K.H. The effect of herpesvirus infection on the expression of cell adhesion molecules on cultured human dermal microvascular endothelial cells. J. Dermatol. Sci. 2000, 24, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Held, K.; Eiglmeier, I.; Himmelein, S.; Sinicina, I.; Brandt, T.; Theil, D.; Dornmair, K.; Derfuss, T. Clonal expansions of CD8þ T cells in latently HSV-1-infected human trigeminal ganglia. J. Neurovirol. 2012, 18, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Savarin, C.; Bergmann, C.C. Viral-induced suppression of self-reactive T cells: Lessons from neurotropic coronavirus-induced demyelination. J. Neuroimmunol. 2017, 308, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Sethi, D.K.; Gordo, S.; Schubert, D.A.; Wucherpfennig, K.W. Cross reactivity of a human autoimmune TCR is dominated by a single TCR loop. Nat. Commun. 2013, 4, 2623. [Google Scholar] [CrossRef]
- Cortese, I.; Capone, S.; Luchetti, S.; Cortese, R.; Nicosia, A. Cross-reactive phage displayed mimotopes lead to the discovery of mimicry between HSV-1 and a brain-specific protein. J. Neuroimmunol. 2001, 113, 119–128. [Google Scholar] [CrossRef]
- Kozin, M.S.; Kulakova, O.G.; Favorova, O.O. Involvement of mitochondria in neurodegeneration in multiple sclerosis. Biochemist 2018, 83, 813–830. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Lewin, A.S.; Sun, L.; Hauswirth, W.W.; Guy, J. Mitochondrial protein nitration primes neurodegeneration in experimental autoimmune encephalomyelitis. J. Biol. Chem. 2006, 281, 31950–31962. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131, 1722–1735. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Le, W. Role of autophagy in the pathogenesis of multiple sclerosis. Neurosci. Bull. 2015, 31, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Wucherpfennig, K.W.; Strominger, J.L. Molecular mimicry in T cell-mediated autoimmunity: Viral peptides activate human T cell clones specific for myelin basic protein. Cell 1995, 80, 695–705. [Google Scholar] [CrossRef]
- Lunemann, J.D.; Jelcić, I.; Roberts, S.; Lutterotti, A.; Tackenberg, B.; Martin, R.; Münz, C. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J. Exp. Med. 2008, 205, 1763–1773. [Google Scholar] [CrossRef]
- Ramasamy, R.; Mohammed, F.; Meier, U.C. HLA DR2b-Binding Peptides From Human Endogenous Retrovirus Envelope, Epstein-Barr Virus and Brain Proteins in the Context of Molecular Mimicry in Multiple Sclerosis. Immunol. Lett. 2020, 217, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Tengvall, K.; Huang, J.; Hellstrom, C.; Kammer, P.; Bistrom, M.; Ayoglu, B.; Bomfim, I.L.; Stridh, P.; Butt, J.; Brenner, N.; et al. Molecular Mimicry Between Anoctamin 2 and Epstein-Barr Virus Nuclear Antigen 1 Associates With Multiple Sclerosis Risk. Proc. Natl. Acad. Sci. USA 2019, 116, 16955–16960. [Google Scholar] [CrossRef] [PubMed]
- van Sechel, A.C.; Bajramovic, J.J.; van Stipdonk, M.J.; Persoon-Deen, C.; Geutskens, S.B.; van Noort, J.M. EBV-induced expression and HLA-DR-restricted presentation by human B cells of alpha B-crystallin, a candidate autoantigen in multiple sclerosis. J. Immunol. 1999, 162, 129–135. [Google Scholar] [CrossRef]
- Lanz, T.V.; brewer, R.C.; Ho, P.P.; Moon, J.S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.S.; Bartley, C.M.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Jelcic, I.; Al Nimer, F.; Wang, J.; Lentsch, V.; Planas, R.; Jelcic, I.; Madjovski, A.; Ruhrmann, S.; Faigle, W.; Frauenknecht, K.; et al. Memory B cells activate brain-homing, autoreactive CD4(+) T cells in multiple sclerosis. Cell 2018, 175, 85–100. [Google Scholar] [CrossRef]
- Nociti, V.; Frisullo, G.; Marti, A.; Luigetti, M.; Iorio, R.; Patanella, A.K.; Bianco, A.; Tonali, P.A.; Grillo, R.L.; Sabatelli, M.; et al. Epstein-Barr virus antibodies in serum and cerebrospinal fluid from multiple sclerosis, chronic inflammatory demyelinating polyradiculoneuropathy and amyotrophic lateral sclerosis. J. Neuroimmunol. 2010, 225, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Kuchroo, V.K.; Weiner, H.L. How does Epstein-Barr virus trigger MS? Immunity 2022, 55, 390–392. [Google Scholar] [CrossRef]
- Castellazzi, M.; Contini, C.; Tamborino, C.; Fasolo, F.; Roversi, G.; Seraceni, S.; Rizzo, R.; Baldi, E.; Tola, M.R.; Bellini, T.; et al. Epstein-Barr virus specific intrathecal oligoclonal IgG production in relapsing-remitting multiple sclerosis is limited to a subset of patients and is composed of low-affinity antibodies. J. Neuroinflamm. 2014, 11, 188. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L.; Lunemann, J.D. The initiation and prevention of multiple sclerosis. Nat. Rev. Neurol. 2012, 8, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Michel, A.; Butt, J.; Brenner, N.; Hillert, J.; Waterboer, T.; Kockum, I.; Olsson, T.; Alfresdsson, L. High levels of Epstein-Barr virus nuclear antigen-1-specific antibodies and infectious mononucleosis act both independently and synergistically to increase multiple sclerosis risk. Front. Neurol. 2019, 10, 1368. [Google Scholar] [CrossRef]
- Disanto, G.; Hall, C.; Lucas, R.; Ponsonby, A.L.; Berlanga-Taylor, A.J.; Giovannoni, G.; the Ausimmune Investigator Group. Assessing Interactions Between HLA-DRB1*15 and Infectious Mononucleosis on the Risk of Multiple Sclerosis. Mult. Scler. 2013, 19, 1355–1358. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.I.; Aziz, K.; Gul, A.; Samar, S.S.; Bareeqa, S.B. Risk of multiple sclerosis in Epstein-Barr virus infection. Cureus 2019, 11, e5699. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Deeba, E.; Koptides, D.; Gaglia, E.; Constantinou, A.; Lambrianides, A.; Pantzaris, M.; Krashias, G.; Christodoulou, C. Evaluation of Epstein-Barr virus-specific antibodies in Cypriot multiple sclerosis patients. Mol. Immunol. 2019, 105, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Baglio, S.R.; van Eijndhoven, M.A.J.; Koppers-Lalic, D.; Berenguer, J.; Lougeed, S.M.; Gibbs, S.; Leveille, N.; Rinkel, R.N.P.M.; Hopmans, E.S.; Swaminathan, S.; et al. Sensing of latent EBV infection through exosomal transfer of 5′pppRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E587–E596. [Google Scholar] [CrossRef] [PubMed]
- Afrasiabi, A.; Fewings, N.L.; Schibeci, S.D.; Keane, J.T.; Booth, D.R.; Parnell, G.P.; Swaminathan, S. The interaction of human and Epstein-Barr virus miRNAs with multiple sclerosis risk loci. Int. J. Mol. Sci. 2021, 22, 2927. [Google Scholar] [CrossRef]
- Absinta, M.; Maric, D.; Gharagozloo, M.; Garton, T.; Smith, M.D.; Jin, J.; Fitzgerald, K.C.; Song, A.; Liu, P.; Lin, J.P.; et al. A Lymphocyte–Microglia–Astrocyte Axis in Chronic Active Multiple Sclerosis. Nature 2021, 597, 709–714. [Google Scholar] [CrossRef]
- Horng, S.; Therattil, A.; Moyon, S.; Gordon, A.; Kim, K.; Argaw, A.T.; Hara, Y.; Marian, J.N.; Sawai, S.; Flodby, P.; et al. Astrocytic Tight Junctions Control Inflammatory CNS Lesion Pathogenesis. J. Clin. Investig. 2017, 127, 3136–3151. [Google Scholar] [CrossRef]
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef]
- Dong, Y.; Yong, V.W. When Encephalitogenic T Cells Collaborate with Microglia in Multiple Sclerosis. Nat. Rev. Neurol. 2019, 15, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Kunkl, M.; Frascolla, S.; Amormino, C.; Volpe, E.; Tuosto, L. T Helper Cells: The Modulators of Inflammation in Multiple Sclerosis. Cells 2020, 9, 482. [Google Scholar] [CrossRef] [PubMed]
- Opsahl, M.L.; Kennedy, P.G.E. Early and late HHV-6 gene transcripts in multiple sclerosis lesions and normal appearing white matter. Brain 2005, 128, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.D.; Mock, D.J.; Powers, J.M.; Baker, J.V.; Blumberg, B.M. Human herpesvirus 6 genome and antigen in acute multiple sclerosis lesions. J. Infect. Dis. 2003, 187, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Pormohammad, A.; Azimi, T.; Falah, F.; Faghihloo, E. Relationship of human herpes virus 6 and multiple sclerosis: A systematic review and meta-analysis. J. Cell Physiol. 2018, 233, 2850–2862. [Google Scholar] [CrossRef]
- Chapenko, S.; Millers, A.; Nora, Z.; Logina, I.; Kukaine, R.; Murovska, M. Correlation Between HHV-6 Reactivation and Multiple Sclerosis Disease Activity. J. Med. Virol. 2003, 69, 111–117. [Google Scholar] [CrossRef]
- Ramroodi, N.; Sanadgol, N.; Ganjali, Z.; Niazi, A.A.; Sarabandi, V.; Moghtaderi, A. Monitoring of active human herpes virus 6 infection in Iranian patients with different subtypes of multiple sclerosis. J. Pathog. 2013, 2013, 194932. [Google Scholar] [CrossRef]
- Bistrom, M.; Jons, D.; Engdahl, E.; Gustafsson, R.; Huang, J.; Brenner, N.; Butt, J.; AlonsoMagdalena, L.; Gunnarsson, M.; Vrethem, M.; et al. Epstein-Barr virus infection after adolescence and human herpesvirus 6A as risk factors for multiple sclerosis. Eur. J. Neurol. 2021, 28, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Engdahl, E.; Gustafsson, R.; Huang, J.; Bistrom, M.; Lima Bomfim, I.; Stridh, P.; Khademi, M.; Brenner, N.; Butt, J.; Michel, A.; et al. Increased serological response against human herpesvirus 6A is associated with risk for multiple sclerosis. Front. Immunol. 2019, 10, 2715. [Google Scholar] [CrossRef]
- Ortega-Madueño, I.; Garcia-Montojo, M.; Dominguez-Mozo, M.I.; GarciaMartinez, A.; Arias-Leal, A.M.; Casanova, I.; Arroyo, R.; Alvarez-Lafuente, R. Anti-Human Herpesvirus 6A/B IgG Correlates with Relapses and Progression in Multiple Sclerosis. PLoS ONE 2014, 9, e104836. [Google Scholar] [CrossRef]
- Soldan, S.S.; Berti, R.; Salem, N.; Secchiero, P.; Flamand, L.; Calabresi, P.A.; Brennan, M.B.; Maloni, H.W.; McFarland, H.F.; Lin, H.C.; et al. Association of Human Herpes Virus 6 (HHV-6) With Multiple Sclerosis: Increased IgM Response to HHV-6 Early Antigen and Detection of Serum HHV-6 DNA. Nat. Med. 1997, 3, 1394–1397. [Google Scholar] [CrossRef]
- Rand, K.H.; Houck, H.; Denslow, N.D.; Heilman, K.M. Epstein-Barr virus nuclear antigen-1 (EBNA-1) associated oligoclonal bands in patients with multiple sclerosis. J. Neurol. Sci. 2000, 173, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Derfuss, T.; Hohlfeld, R.; Meinl, E. Intrathecal antibody (IgG) production against human herpesvirus type 6 occurs in about 20% of multiple sclerosis patients and might be linked to a polyspecific B-cell response. J. Neurol. 2005, 252, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Tejada-Simon, M.V.; Zang, Y.C.; Hong, J.; Rivera, V.M.; Zhang, J.Z. Cross-Reactivity with Myelin Basic Protein and Human Herpesvirus-6 in Multiple Sclerosis. Ann. Neurol. 2003, 53, 189–197. [Google Scholar] [CrossRef]
- Cheng, W.; Ma, Y.; Gong, F.; Hu, C.; Qian, L.; Huang, Q.; Yu, Q.; Zhang, J.; Chen, S.; Liu, Z.; et al. Cross-reactivity of autoreactive T cells with MBP and viral antigens in patients with MS. Front. Biosci. 2012, 17, 1648–1658. [Google Scholar] [CrossRef]
- Mayne, M.; Cheadle, C.; Soldan, S.S.; Cermelli, C.; Yamano, Y.; Akhyani, N.; Nagel, J.E.; Taub, D.D.; Becker, K.G.; Jacobson, S. Gene expression profile of herpesvirus infected T cells obtained using immune microarrays: Induction of proinflammatory mechanisms. J. Virol. 2001, 75, 11641–11650. [Google Scholar] [CrossRef]
- Dietrich, J.; Blumberg, B.M.; Roshal, M.; Baker, J.V.; Hurley, S.D.; Mayer-Proschel, M.; Mock, D.J. Infection with an endemic human herpesvirus disrupts critical glial precursor cell properties. J. Neurosci. 2004, 24, 4875–4883. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.M.; Gallo, V. The translational biology of remyelination: Past, present, and future. Glia 2014, 62, 1905–1915. [Google Scholar] [CrossRef]
- Cuomo, L.; Angeloni, A.; Zompetta, C.; Cirone, M.; Calogero, A.; Frati, L.; Ragona, G.; Faggioni, A. Human Herpesvirus 6 Variant A, But Not Variant B, Infects EBV-Positive B Lymphoid Cells, Activating the Latent EBV Genome Through a BZLF-1-Dependent Mechanism. AIDS Res. Hum. Retroviruses 1995, 11, 1241–1245. [Google Scholar] [CrossRef]
- Turcanova, V.L.; Bundgaard, B.; Hollsberg, P. Human Herpesvirus-6B Induces Expression of the Human Endogenous Retrovirus K18-Encoded Superantigen. J. Clin. Virol. 2009, 46, 15–19. [Google Scholar] [CrossRef]
- Soldan, S.S.; Fogdell-Hahn, A.; Brennan, M.B.; Mittleman, B.B.; Ballerini, C.; Massacesi, L.; Seya, T.; McFarland, H.F.; Jacobson, S. Elevated serum and cerebrospinal fluid levels of soluble human herpesvirus type 6 cellular receptor, membrane cofactor protein, in patients with multiple sclerosis. Ann. Neurol. 2001, 50, 486–493. [Google Scholar] [CrossRef]
- Sanadgol, N.; Ramroodi, N.; Ahmadi, G.A.; Komijani, M.; Moghtaderi, A.; Bouzari, M.; Rezaei, M.; Kardi, M.T.; Dabiri, S.; Moradi, M.; et al. Prevalence of Cytomegalovirus Infection and Its Role in Total Immunoglobulin Pattern in Iranian Patients with Different Subtypes of Multiple Sclerosis. New Microbiol. 2011, 34, 263–274. [Google Scholar] [PubMed]
- Najafi, S.; Ghane, M.; Poortahmasebi, V.; Jazayeri, S.M.; Yousefzadeh-Chabok, S. Prevalence of Cytomegalovirus in Patients with Multiple Sclerosis: A Case-Control Study in Northern Iran. Jundishapur. J. Microbiol. 2016, 9, e36582. [Google Scholar] [CrossRef] [PubMed]
- Smyk, D.S.; Alexander, A.K.; Walker, M.; Walker, M. Acute disseminated encephalomyelitis progressing to multiple sclerosis: Are infectious triggers involved? Immunol. Res. 2014, 60, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Vanheusden, M.; Stinissen, P.; ’t Hart, B.A.; Hellings, N. Cytomegalovirus: A Culprit or Protector in Multiple Sclerosis? Trends Mol. Med. 2015, 21, 16–23. [Google Scholar] [CrossRef]
- Clerico, M.; De Mercanti, S.; Artusi, C.A.; Durelli, L.; Naismith, R.T. Active CMV Infection in Two Patients with Multiple Sclerosis Treated with Alemtuzumab. Mult. Scler. 2017, 23, 874–876. [Google Scholar] [CrossRef]
- Zheng, M.M.; Zhang, X.H. Cross-reactivity between human cytomegalovirus peptide 981-1003 and myelin oligodendroglia glycoprotein peptide 35-55 in experimental autoimmune encephalomyelitis in Lewis rats. Biochem. Biophys. Res. Commun. 2014, 443, 1118–1123. [Google Scholar] [CrossRef]
- Maple, P.A.C.; Tanasescu, R.; Gran, B.; Constantinescu, C.S. A Different Response to Cytomegalovirus (CMV) and Epstein-Barr Virus (EBV) Infection in UK People with Multiple Sclerosis (PwMS) Compared to Controls. J. Infect. 2020, 80, 320–325. [Google Scholar] [CrossRef]
- Zabalza, A.; Vera, A.; Alari-Pahissa, E.; Munteis, E.; Moreira, A.; Yélamos, J.; Llop, M.; López-Botet, M.; Martínez-Rodríguez, J.E. Impact of Cytomegalovirus Infection on B Cell Differentiation and Cytokine Production in Multiple Sclerosis. J. Neuroinflamm. 2020, 17, 161. [Google Scholar] [CrossRef]
- Kang, J.-H.; Sheu, J.-J.; Kao, S.; Lin, H.C. Increased risk of multiple sclerosis following herpes zoster: A nationwide, population-based study. J. Infect. Dis. 2011, 204, 188–192. [Google Scholar] [CrossRef]
- Jarius, S.; Eichhorn, P.; Franciotta, D.; Petereit, H.F.; Akman-Demir, G.; Wick, M.; Wildemann, B. The MRZ reaction as a highly specific marker of multiple sclerosis: Re-evaluation and structured review of the literature. J. Neurol. 2017, 264, 453–466. [Google Scholar] [CrossRef]
- Rice, E.M.; Thakolwiboon, S.; Avila, M. Geographic heterogeneity in the association of varicella-zoster virus seropositivity and multiple sclerosis: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2021, 53, 103024. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Ordoñez, G.; Pineda, B.; Flores, J. The participation of varicella zoster virus in relapses of multiple sclerosis. Clin. Neurol. Neurosurg. 2014, 119, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Zhou, S.; Xu, Y.; Gao, R.; Zhang, M.; Zeng, Q.; Su, W.; Wang, R. Chickenpox and multiple sclerosis: A Mendelian randomization study. J. Med. Virol. 2022, 95, e28315. [Google Scholar] [CrossRef]
- Moon, U.Y.; Park, S.J.; Oh, S.T.; Kim, W.U.; Park, S.H.; Lee., S.H.; Choo, C.S.; Kim, H.Y.; Lee, W.K.; Leel, S.K. Patients with systemic lupus erythematosus have abnormally elevated Epstein-Barr virus load in blood. Arthritis Res. Ther. 2004, 6, R295–R302. [Google Scholar] [CrossRef]
- Chen, X.; Li, H.; Wu, C.; Zhang, Y. Epstein‒Barr virus and human herpesvirus 6 infection in patients with systemic lupus erythematosus. Virol. J. 2023, 20, 29. [Google Scholar] [CrossRef] [PubMed]
- Berkun, Y.; Zandman-Goddard, G.; Barzilai, O.; Boaz, M.; Sherer, Y.; Larida, B.; Blank, M.; Anaya, J.M.; Shoenfeld, Y. Infectious antibodies in systemic lupus erythematosus patients. Lupus 2009, 18, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- James, J.A.; Neas, B.R.; Moser, K.L.; Hall, T.; Bruner, G.R.; Sestak, A.L.; Harley, J.B. Systemic lupus erythematosus in adults is associated with previous Epstein-Barr virus exposure. Arthritis Rheum. 2001, 44, 1122–1126. [Google Scholar] [CrossRef]
- Chougule, D.; Nadkar, M.; Rajadhyaksha, A.; Khadilkar, P.; Patwardhan, M.; Kaveri, S.; Ghosh, K.; Pradhan, V. Association of clinical and serological parameters of systemic lupus erythematosus patients with Epstein-Barr virus antibody profile. J. Med. Virol. 2018, 90, 559–563. [Google Scholar] [CrossRef]
- Jog, N.R.; Young, K.A.; Munroe, M.E.; Harmon, M.T.; Guthridge, J.M.; Kelly, J.A.; Kame, D.L.; Gilkeson, G.S.; Weisman, M.H.; Karp, D.R.; et al. Association of Epstein-Barr virus serological reactivation with transitioning to systemic lupus erythematosus in at-risk individuals. Ann. Rheum. Dis. 2019, 78, 1235–1241. [Google Scholar] [CrossRef]
- James, J.A.; Harley, J.B.; Scofield, R.H. Epstein-Barr virus and systemic lupus erythematosus. Curr. Opin. Rheumatol. 2006, 18, 462–467. [Google Scholar] [CrossRef]
- Yadav, P.; Tran, H.; Ebegbe, R.; Gottlieb, P.; Wei, H.; Lewis, R.H.; Mumbey-Wafula, A.; Kaplan, A.; Kholdarova, E.; Spatz, L. Antibodies elicited in response to EBNA-1 may cross-react with dsDNA. PLoS ONE 2011, 6, e14488. [Google Scholar] [CrossRef] [PubMed]
- Csorba, K.; Schirmbeck, L.A.; Tuncer, E.; Ribi, C.; Roux-Lombard, P.; Chizzolini, C.; Huynh-Do, U.; Vanhecke, D.; Trendelenburg, M. Anti-C1q Antibodies as Occurring in Systemic Lupus Erythematosus Could Be Induced by an Epstein-Barr Virus-Derived Antigenic Site. Front. Immunol. 2019, 10, 2619. [Google Scholar] [CrossRef]
- Incaprera, M.; Rindi, L.; Bazzichi, A.; Garzelli, C. Potential role of the Epstein– Barr virus in systemic lupus erythematosus autoimmunity. Clin. Exp. Rheumatol. 1998, 16, 289–294. [Google Scholar]
- Tu, J.; Wang, X.; Geng, G.; Xue, X.; Lin, X.; Zhu, X.; Sun, L. The Possible Effect of B-Cell Epitopes of Epstein-Barr Virus Early Antigen, Membrane Antigen, Latent Membrane Protein-1, and -2A on Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 187. [Google Scholar] [CrossRef]
- Jog, N.R.; James, J.A. Epstein Barr Virus and Autoimmune Responses in Systemic Lupus Erythematosus. Front. Immunol. 2021, 11, 623944. [Google Scholar] [CrossRef]
- Jog, N.R.; Chakravarty, E.F.; Guthridge, J.M.; James, J.A. Epstein Barr Virus Interleukin 10 Suppresses Anti-inflammatory Phenotype in Human Monocytes. Front. Immunol. 2018, 9, 2198. [Google Scholar] [CrossRef]
- Stewart, J.P.; Rooney, C.M. The interleukin-10 homolog encoded by Epstein-Barr virus enhances the reactivation of virus-specific cytotoxic T cell and HLA unrestricted killer cell responses. Virology 1992, 191, 773–782. [Google Scholar] [CrossRef]
- HoHsieh, A.; Wang, C.M.; Wu, Y.-J.J.; Chen, A.; Chang, M.-I.; Chen, J.-Y. B Cell Epitope of Human Cytomegalovirus Phosphoprotein 65 (HCMV Pp65) Induced Anti-DsDNA Antibody in BALB/c Mice. Arthritis Res. Ther. 2017, 19, 65. [Google Scholar] [CrossRef]
- Hsieh, A.-H.; Kuo, C.-F.; Chou, I.-J.; Tseng, W.-Y.; Chen, Y.-F.; Yu, K.-H.; Luo, S.-F. Human Cytomegalovirus Pp65 Peptide-Induced Autoantibodies Cross-Reacts with TAF9 Protein and Induces Lupus-like Autoimmunity in BALB/c Mice. Sci. Rep. 2020, 10, 9662. [Google Scholar] [CrossRef]
- Neo, J.Y.J.; Wee, S.Y.K.; Bonne, I.; Tay, S.H.; Raida, M.; Jovanovic, V.; Fairhurst, A.-M.; Lu, J.; Hanson, B.J.; MacAry, P.A. Characterisation of a Human Antibody That Potentially Links Cytomegalovirus Infection with Systemic Lupus Erythematosus. Sci. Rep. 2019, 9, 9998. [Google Scholar] [CrossRef]
- Guo, G.; Ye, S.; Xie, S.; Ye, L.; Lin, C.; Yang, M.; Shi, X.; Wang, F.; Li, B.; Li, M.; et al. The Cytomegalovirus Protein US31 Induces Inflammation through Mono-Macrophages in Systemic Lupus Erythematosus by Promoting NF-KB2 Activation. Cell Death Dis. 2018, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Rozenblyum, E.V.; Allen, U.D.; Silverman, E.D.; Levy, D.M. Cytomegalovirus Infection in Childhood-Onset Systemic Lupus Erythematosus. Int. J. Clin. Rheumatol. 2013, 8, 137–146. [Google Scholar] [CrossRef]
- Janahi, E.M.A.; Das, S.; Bhattacharya, S.N.; Haque, S.; Akhter, N.; Jawed, A.; Wahid, M.; Mandal, R.K.; Lohani, M.; Areeshi, M.Y.; et al. Cytomegalovirus Aggravates the Autoimmune Phenomenon in Systemic Autoimmune Diseases. Microb. Pathog. 2018, 120, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Kivity, S.; Arango, M.T.; Ehrenfeld, M.; Tehori, O.; Shoenfeld, Y.; Anaya, J.M.; Agmon-Levin, N. Infection and autoimmunity in Sjogren’s syndrome: A clinical study and comprehensive review. J. Autoimmun. 2014, 51, 17–22. [Google Scholar] [CrossRef]
- Sanosyan, A.; Daien, C.; Nutz, A.; Bollore, K.; Bedin, A.S.; Morel, J.; Zimmermann, V.; Nocturne, G.; Peries, M.; Guigue, N.; et al. Discrepancy of Serological and Molecular Patterns of Circulating Epstein-Barr Virus Reactivation in Primary Sjögren’s Syndrome. Front. Immunol. 2019, 10, 1153. [Google Scholar] [CrossRef]
- Barcelos, F.; Martins, C.; Monteiro, R.; Cardigos, J.; Prussiani, T.; Sitima, M.; Alves, N.; Vaz-Patto, J.; Cunha-Branco, J.; Borrego, L.M. Association between EBV serological patterns and lymphocytic profle of SjS patients support a virally triggered autoimmune epithelitis. Sci. Rep. 2021, 11, 4082. [Google Scholar] [CrossRef] [PubMed]
- Saito, I.; Servenius, B.; Compton, T.; Fox, R.I. Detection of Epstein-Barr virus DNA by polymerase chain reaction in blood and tissue biopsies from patients with Sjögren’s syndrome. J. Exp. Med. 1989, 169, 2191–2198. [Google Scholar] [CrossRef] [PubMed]
- Casanova, J.L.; Abel, L. Mechanisms of viral inflammation and disease in humans. Science 2021, 374, 1080–1086. [Google Scholar] [CrossRef]
- Hou, X.; Hong, X.; Ou, M.; Meng, S.; Wang, T.; Liao, S.; He, J.; Yu, H.; Liu, L.; Yin, L.; et al. Analysis of Gene Expression and TCR/B Cell Receptor Profiling of Immune Cells in Primary Sjogren’s Syndrome by Single-Cell Sequencing. J. Immunol. 2022, 209, 238–249. [Google Scholar] [CrossRef]
- Maslinska, M. The role of Epstein-Barr virus infection in primary Sjogren’s syndrome. Curr. Opin. Rheumatol. 2019, 31, 475–483. [Google Scholar] [CrossRef]
- Huang, P.J.; Lin, S.P.; Wu, C.Y.; Liu, Y.T.; Chen, H.H. Association between a History of herpes zoster and the risk of Sjögren’s syndrome: A nationwide, population-based, case– control study. BMJ Open 2022, 12, e061962. [Google Scholar] [CrossRef]
- Newkirk, M.M.; Duffy, K.N.W.; Lecrerc, J.; Lambert, N.; Shiroky, J.B. Detection of cytomegalovirus, Epstein-Barr and herpes virus—6 in patients with rheumatoid arthritis with or without sjogren’s syndrome. Rheumatology 1994, 33, 317–322. [Google Scholar] [CrossRef]
- Broccolo, F.; Fusetti, L.; Ceccherini-Nelli, L. Possible role of human herpesvirus 6 as a trigger of autoimmune disease. Sci. World J. 2013, 24, 867389. [Google Scholar] [CrossRef]
- Takizawa, Y.; Inokuma, S.; Tanaka, Y.; Saito, K.; Atsumi, T.; Hirakata, M.; Kameda, H.; Hirohata, S.; Kondo, H.; Kumagai, S.; et al. Clinical characteristics of cytomegalovirus infection in rheumatic diseases: Multicentre survey in a large patient population. Rheumatology 2008, 47, 1373–1378. [Google Scholar] [CrossRef]
- Leonard, L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus exposure and neurodegenerative disease risk across national biobanks. Neuron 2023, 111, 1086–1093. [Google Scholar]
- Itzaki, R.F.; Tabet, N. Herpes simplex encephalitis and Alzheimer’s disease: Is there a link? J. Neurol. Sci. 2017, 380, 20–21. [Google Scholar] [CrossRef]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Li Puma, D.D.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes simplex virus-1 in the brain: The dark side of a sneaky infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef]
- Huang, S.Y.; Yang, Y.X.; Kuo, K.; Li, H.Q.; Shen, X.N.; Chen, S.D.; Cui, M.; Tan, L.; Dong, J.T.; Yu, J.T. Herpesvirus infections and Alzheimer’s disease: A Mendelian randomization study. Alzheimers Res. Ther. 2021, 13, 158. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, L.J.; Yang, L.; Yang, C.S.; Yi, M.; Zhang, S.N.; Wang, N.; Huang, C.N.; Liu, M.Q. Positive association of herpes simplex virus-IgG with multiple sclerosis: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2021, 47, 102633. [Google Scholar] [CrossRef]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef]
- Fominykh, V.; Shadrin, A.A.; Jaholkowski, P.P.; Bahrami, S.; Athanasiu, L.; Wightman, D.P.; Uffelmann, E.; Posthuma, D.; Selbaek, G.; Dale, A.M.; et al. Shared genetic loci between Alzheimer’s disease and multiple sclerosis: Crossroads between neurodegeneration and immune system. Neurobiol. Dis. 2023, 183, 106174. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, U.H.; Zeng, E.; Pasinetti, G.M. The use of antimicrobial and antiviral drugs in Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 4920. [Google Scholar] [CrossRef] [PubMed]
- Hui, Z.; Zhijun, Y.; Yushan, Y.; Liping, C.; Yiying, Z.; Difan, Z.; Chunglit, C.T.; Wei, C. The combination of acyclovir and dexamethasone protects against Alzheimer’s disease-related cognitive impairments in mice. Psychopharmacology 2020, 237, 1851–1860. [Google Scholar] [CrossRef]
- Schnier, C.; Janbek, J.; Lathe, R.; Haas, J. Reduced dementia incidence after Varicella zoster vaccination in Wales 2013–2020. Alzheimers Dement. 2022, 8, e12293. [Google Scholar] [CrossRef] [PubMed]
- Scherrer, J.F.; Salas, J.; Wiemken, T.L.; Hoft, D.F.; Jacobs, C.; Morley, J.E. Impact of herpes zoster vaccination on incident dementia: A retrospective study in two patient cohorts. PLoS ONE 2021, 16, e0257405. [Google Scholar] [CrossRef]
- Lehrer, S.; Rheinstein, P.H. Herpes zoster vaccination reduces risk of dementia. In Vivo 2021, 35, 3271–3275. [Google Scholar] [CrossRef]
- Lophatananon, A.; Mekli, K.; Cant, R.; Burns, A.; Dobson, C.; Itzhaki, R.; Muir, K. Shingles, Zostavax vaccination and risk of developing dementia: A nested case–control study—Results from the UK Biobank cohort. BMJ Open 2021, 11, e045871. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Result | Study Type | Refs |
|---|---|---|---|
| AD | The presence of Aβ and HSV-1 cross-reactive antibodies in patients with AD | Comparative study | [73] |
| An increased level of IgM antibodies against HSV-1,2 was associated with an increased risk of developing dementia | Prospective cohort studies | [74,75] | |
| AD-derived B lymphocytes infected with EBV produced high levels of TNF-α in vitro | Cell model studies | [78,79] | |
| EBV antigens stimulated cytotoxic and proinflammatory functions by CD45RA+ cells | Prospective cohort and knockout studies | [80,81] | |
| Increased CMV blood markers associated with cognitive decline | Longitudinal, prospective cohort studies | [76,100] | |
| Levels of CMV IgG antibodies associated with increased neurofibrillary tangles | Longitudinal study | [101] | |
| Patients with herpes zoster showed a significantly increased risk of developing AD | Retrospective cohort studies | [95,96] | |
| Studies showed an association between CMV and moderate–severe dementia | Population cohort study | [99] | |
| HSV-1 induced neuroinflammation | In vitro model studies | [47,48,49,50,51] | |
| HSV-1 induced the production of both Aβ and the tau protein in human neural cells | In vitro infection studies | [55] | |
| HSV-1 DNA was present in the brain of AD patients | Molecular studies | [57,58] | |
| EBV was present in the blood and brains of AD patients | Comparative and molecular study | [76] | |
| EBV reactivation has been associated with cognitive decline | Longitudinal study | [77] | |
| EBV BNLF-2a has been associated with AD progression | Biochemical studies | [82,83] | |
| HHV-6-infected microglia showed an accumulation of Aβ and tau proteins | In vivo and in vitro infection model | [85,86] | |
| HHV-6 reduced autophagy | In vitro infection model | [87,88,89] | |
| HHV-6-infected patients and AD patients shared 95 differentially expressed genes | Computational analysis study | [90] | |
| Binding of VZV to the insulin-degrading enzyme | In vitro infection model | [97,98] | |
| PD | Molecular mimicry between HSV-1 and α-synuclein | Seroprevalence study | [102] |
| Molecular mimicry between EBV LMP1 and α-synuclein | Seroprevalence study | [104] | |
| CMV-, EBV-, and HHV-6-infected patients shared several differentially expressed genes with AD patients | Computational analysis study | [90] |
| Disease | Result | Study Type | Refs |
|---|---|---|---|
| MS | Molecular mimicry between HSV-1 protein and myelin basic protein | Biochemical study | [150] |
| Molecular mimicry between EBV protein LMP-1 and different proteins involved in MS pathogenesis | Case–control studies | [152,153,154,155] | |
| Molecular mimicry between EBV proteins BHRF1 and BPLF1 with a protein present in neurons | Epitope discover approach and cell immunity analysis | [158] | |
| Defective cytotoxic T cells control of EBV in MS | Seroprevalence study | [159] | |
| Oligoclonal IgG against EBV have been detected in the brain of MS patients | Comparative study | [161] | |
| Higher titers of EBV antibodies have been detected in MS patients | Case control study | [164] | |
| Serological response against HHV-6 was higher in MS patients | Molecular, case–control studies | [179,180,181,182,183] | |
| Oligoclonal IgG against HHV-6 have been detected in the brains of MS patients | Seroprevalence study | [184] | |
| Molecular mimicry between HHV-6 protein U24 and myelin basic protein | Seroprevalence study | [186] | |
| Increased level of sCD46, the receptor for HHV-6 has been detected in MS patients | Immunological study | [193] | |
| Molecular mimicry between HCMV antigen (UL86981-1003) and myelin oligodendrocyte glycoprotein (MOG) | In vivo experimental model | [199] | |
| Patients with herpes zoster have been associated with a higher risk of MS | Population-based study and computational GWAS | [202,206] | |
| HSV-1 DNA in the blood and in the brains of MS patients | Molecular studies | [138,139] | |
| HSV-1 infection-induced CNS demyelination and neuroinflammation | In vitro and in vivo infection studies | [140,141,142,143,145] | |
| EBV-induced an inflammatory environment | In vitro infection and molecular studies | [168,169,170,171,172,173] | |
| High load of HCMV genome has been detected in MS patients | Case–control study | [195] | |
| High load of VZV genome has been detected in MS patients | Molecular study | [205] | |
| SLE | Serological response against EBV was higher in SLE patients | Case–control studies | [209,210,211] |
| EBV antibodies cross-reacted with autoantigens of SLE | Seroprevalence studies | [215,216,217] | |
| Similarity between CMV antigens and autoantigens of SLE | Animal model and in vitro infection studies | [222,223] | |
| High load of EBV DNA was detected in SLE patients | Molecular study | [207] | |
| vIL-10, a viral EBV protein, has been detected at higher concentrations in plasma of SLE patients | In vitro infection model | [219] | |
| SS | Higher titers of anti-HHV-6 have been found in a group of SS patients | Seroprevalence study | [235] |
| A significant correlation between herpes zoster exposure and SS risk has been detected | Population-based case–control study | [234] | |
| High levels of EBV DNA have been detected in SS patients | Molecular study | [230] | |
| Increased EBV reactivation has been observed in SS patients | Observational study | [228] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Francesco, M.A. Herpesviridae, Neurodegenerative Disorders and Autoimmune Diseases: What Is the Relationship between Them? Viruses 2024, 16, 133. https://doi.org/10.3390/v16010133
De Francesco MA. Herpesviridae, Neurodegenerative Disorders and Autoimmune Diseases: What Is the Relationship between Them? Viruses. 2024; 16(1):133. https://doi.org/10.3390/v16010133
Chicago/Turabian StyleDe Francesco, Maria Antonia. 2024. "Herpesviridae, Neurodegenerative Disorders and Autoimmune Diseases: What Is the Relationship between Them?" Viruses 16, no. 1: 133. https://doi.org/10.3390/v16010133