
Successful Confirmation of Dual Genital Herpes Co-Infection with Herpes Simplex Virus 1 and Herpes Simplex Virus 2 Using Unbiased Metagenomic Next-Generation Sequencing
 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
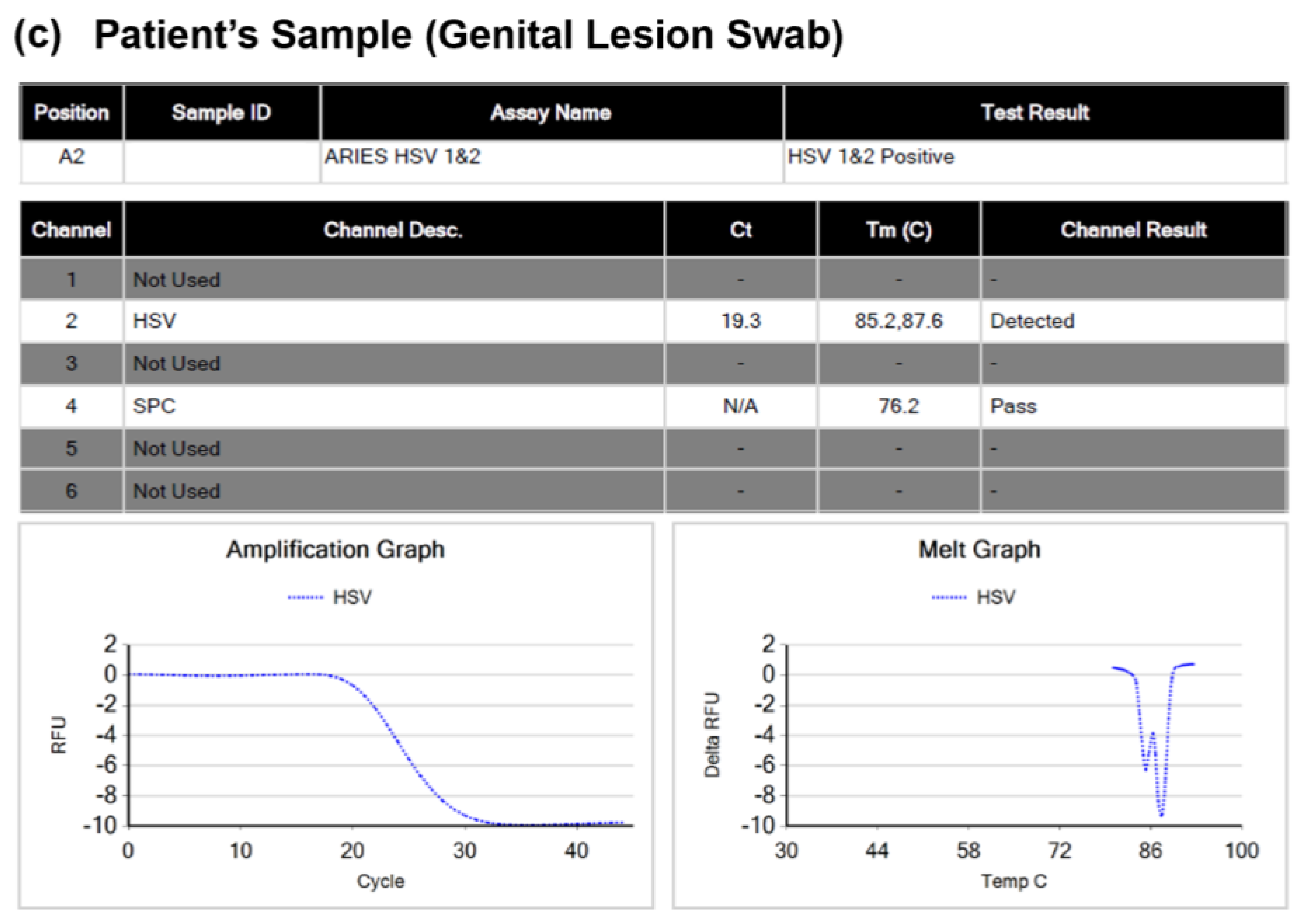
3.1. Luminex ARIES HSV 1&2 Assay
3.2. Unbiased Metagenomic-Based Next-Generation Sequencing (mNGS)
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gupta, R.; Warren, T.; Wald, A. Genital herpes. Lancet 2007, 370, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, D.W.; Rouse, D.J. Clinical practice. Genital herpes. N. Engl. J. Med. 2004, 350, 1970–1977. [Google Scholar] [CrossRef] [PubMed]
- Corey, L.; Wald, A. Maternal and neonatal herpes simplex virus infections. N. Engl. J. Med. 2009, 361, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Namvar, L.; Olofsson, S.; Bergström, T.; Lindh, M. Detection and typing of herpes simplex virus (HSV) in mucocutaneous samples by TaqMan PCR targeting a gB segment homologous for HSV types 1 and 2. J. Clin. Microbiol. 2005, 43, 2058–2064. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Chai, C.N.; Capinpin, S.M.; Ang, A.; Ng, S.Y.; Lee, P.L.; Ng, C.W.S.; Yan, G.; Lee, H.K.; Chiu, L.L.; et al. Evaluation of the Luminex ARIES HSV 1&2 assay and comparison with the FTD Neuro 9 and In-house Real-Time PCR assays for detecting herpes simplex viruses. Ann. Lab. Med. 2018, 38, 440–445. [Google Scholar] [CrossRef]
- AMP Case Report: Identification of Encephalomyocarditis Virus Using Metagenomic NGS in a Patient with Acute Febrile Illness. Available online: https://www.captodayonline.com/amp-case-report-identification-of-encephalomyocarditis-virus-using-metagenomic-ngs-in-a-patient-with-acute-febrile-illness/ (accessed on 29 March 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Lunter, G.; Goodson, M. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011, 21, 936–939. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Lamers, S.L.; Newman, R.M.; Laeyendecker, O.; Tobian, A.A.R.; Colgrove, R.C.; Ray, S.C.; Koelle, D.M.; Cohen, J.; Knipe, D.M.; Quinn, T.C. Global Diversity within and between Human Herpesvirus 1 and 2 Glycoproteins. J. Virol. 2015, 89, 8206–8218. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.A.; Barrell, B. Artemis: Sequence Visualization and Annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.; Chong, H.; Irvine, B.; Domagalski, J. Genital Co-Infection with Herpes Simplex Viruses Type 1 and 2: Comparison of Real-Time PCR Assay and Traditional Viral Isolation Methods. J. Cell. Mol. Med. 2007, 11, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Casto, A.M.; Roychoudhury, P.; Xie, H.; Selke, S.; Perchetti, G.A.; Wofford, H.; Huang, M.; Verjans, G.M.G.M.; Gottlieb, G.S.; Wald, A.; et al. Large, Stable, Contemporary Interspecies Recombination Events in Circulating Human Herpes Simplex Viruses. J. Infect. Dis. 2020, 221, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Heller, M.; Dix, R.D.; Baringer, J.R.; Schachter, J.; Conte, J.E., Jr. Herpetic proctitis and meningitis: Recovery of two strains of herpes simplex virus type 1 from cerebrospinal fluid. J. Infect. Dis. 1982, 146, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.W.; Sistrunk, W.; Binnicker, M.J. Simultaneous detection of herpes simplex virus 1 and 2 in the cerebrospinal fluid of a patient with seizures and encephalitis. J. Clin. Microbiol. 2015, 53, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, A.; Martic, J.; Stanojevic, M.; Jankovic, S.; Nedeljkovic, J.; Nikolic, L.; Pasic, S.; Jankovic, B.; Jovanovic, T. Disseminated neonatal herpes caused by herpes simplex virus types 1 and 2. Emerg. Infect. Dis. 2007, 13, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Fujie Xu, F.; Sternberg, M.R.; Kottiri, B.J.; McQuillan, G.M.; Lee, F.K.; Nahmias, A.J.; Berman, S.M.; Markowitz, L.E. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 2006, 296, 964–973. [Google Scholar] [CrossRef]
- Beydoun, H.A.; Dail, J.; Ugwu, B.; Boueiz, A.; Beydoun, M.A. Socio-demographic and behavioral correlates of herpes simplex virus type 1 and 2 infections and co-infections among adults in the USA. Int. J. Infect. Dis. 2010, 14, e154–e160. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.K.; Ng, S.Y.; Chai, C.N.; Lim, Y.F.; Hu, T.J.; Lee, O.F.; Yan, G. Successful Confirmation of Dual Genital Herpes Co-Infection with Herpes Simplex Virus 1 and Herpes Simplex Virus 2 Using Unbiased Metagenomic Next-Generation Sequencing. Viruses 2023, 15, 1957. https://doi.org/10.3390/v15091957
Lee CK, Ng SY, Chai CN, Lim YF, Hu TJ, Lee OF, Yan G. Successful Confirmation of Dual Genital Herpes Co-Infection with Herpes Simplex Virus 1 and Herpes Simplex Virus 2 Using Unbiased Metagenomic Next-Generation Sequencing. Viruses. 2023; 15(9):1957. https://doi.org/10.3390/v15091957
Chicago/Turabian StyleLee, Chun Kiat, Sau Yoke Ng, Chean Nee Chai, Yu Feng Lim, Tiffany Jingyan Hu, Ogestelli Fabia Lee, and Gabriel Yan. 2023. "Successful Confirmation of Dual Genital Herpes Co-Infection with Herpes Simplex Virus 1 and Herpes Simplex Virus 2 Using Unbiased Metagenomic Next-Generation Sequencing" Viruses 15, no. 9: 1957. https://doi.org/10.3390/v15091957