
Characterization of Diverse Anelloviruses, Cressdnaviruses, and Bacteriophages in the Human Oral DNA Virome from North Carolina (USA)
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Viral Nucleic Acid Extraction, Sequencing, De Novo Assembly, and Virus Genome Identification
2.3. Distribution of Virus Genomes across the Samples
2.4. Phylogenetic Analyses
2.4.1. Anelloviruses
2.4.2. Cressdnaviruses
2.4.3. Microviruses
2.4.4. Large Bacteriophages and Inoviruses
3. Results and Discussion
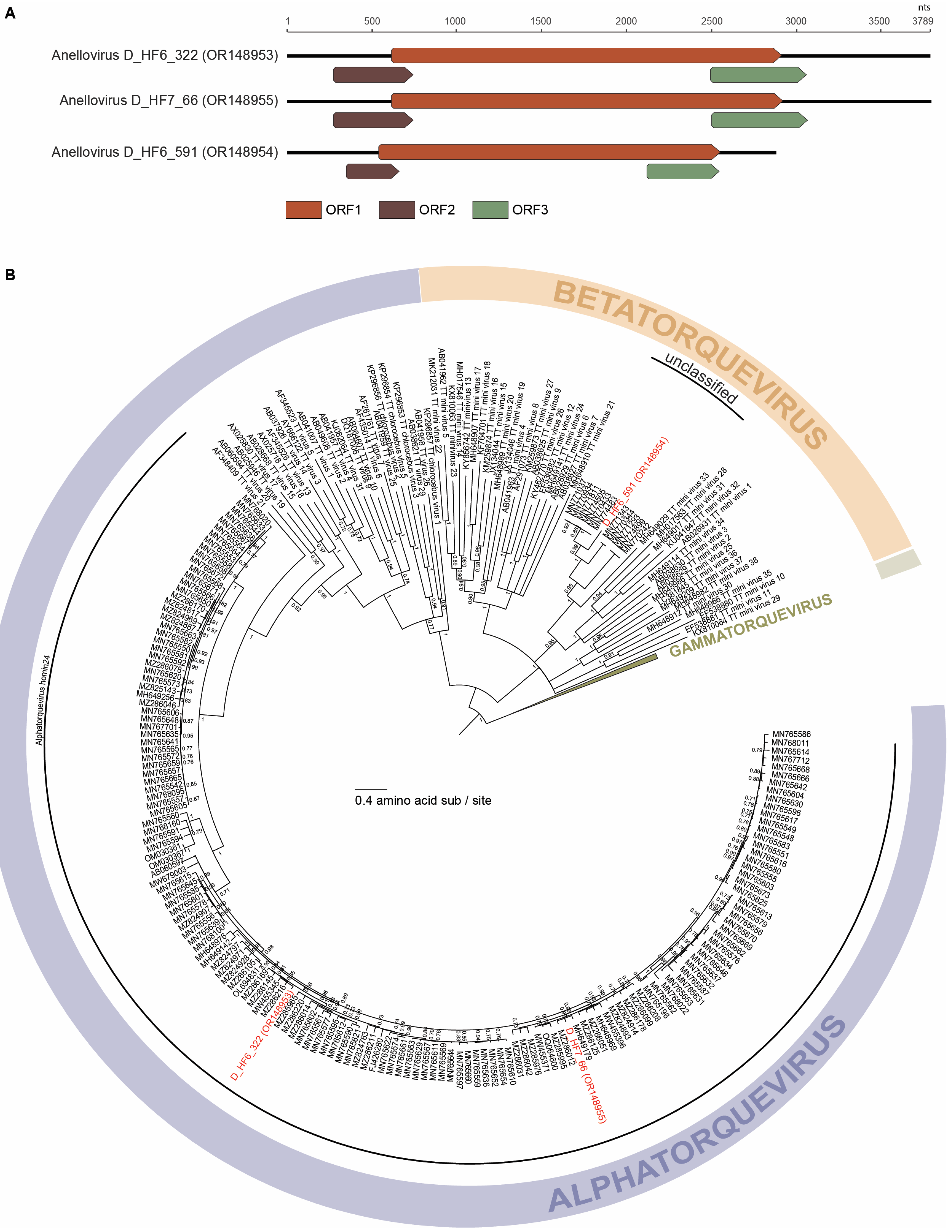
3.1. Anelloviruses
3.2. Cressdnaviruses
3.2.1. Redondoviruses
3.2.2. Unclassified Cressdnaviruses
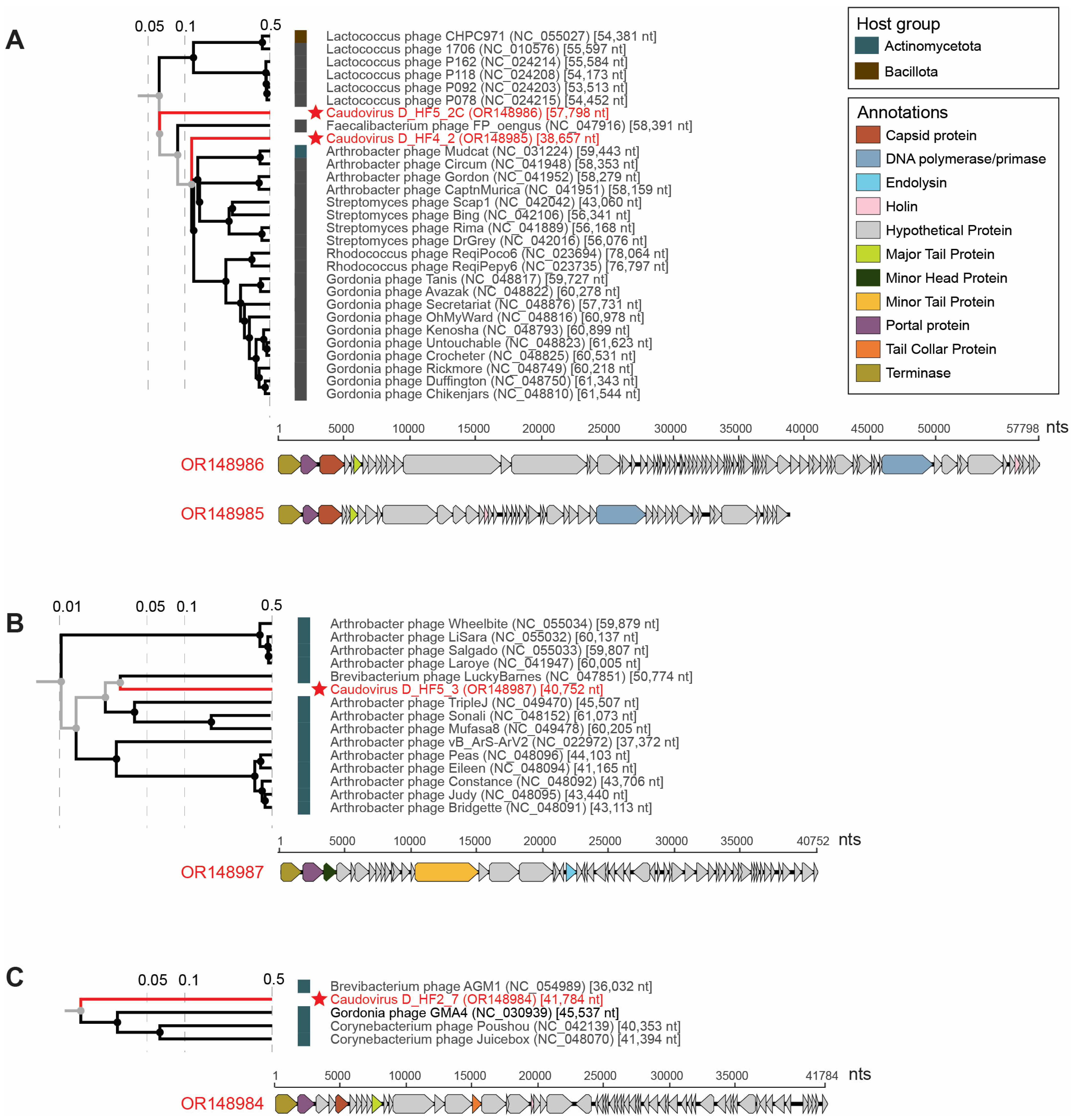
3.3. Tailed dsDNA Phages
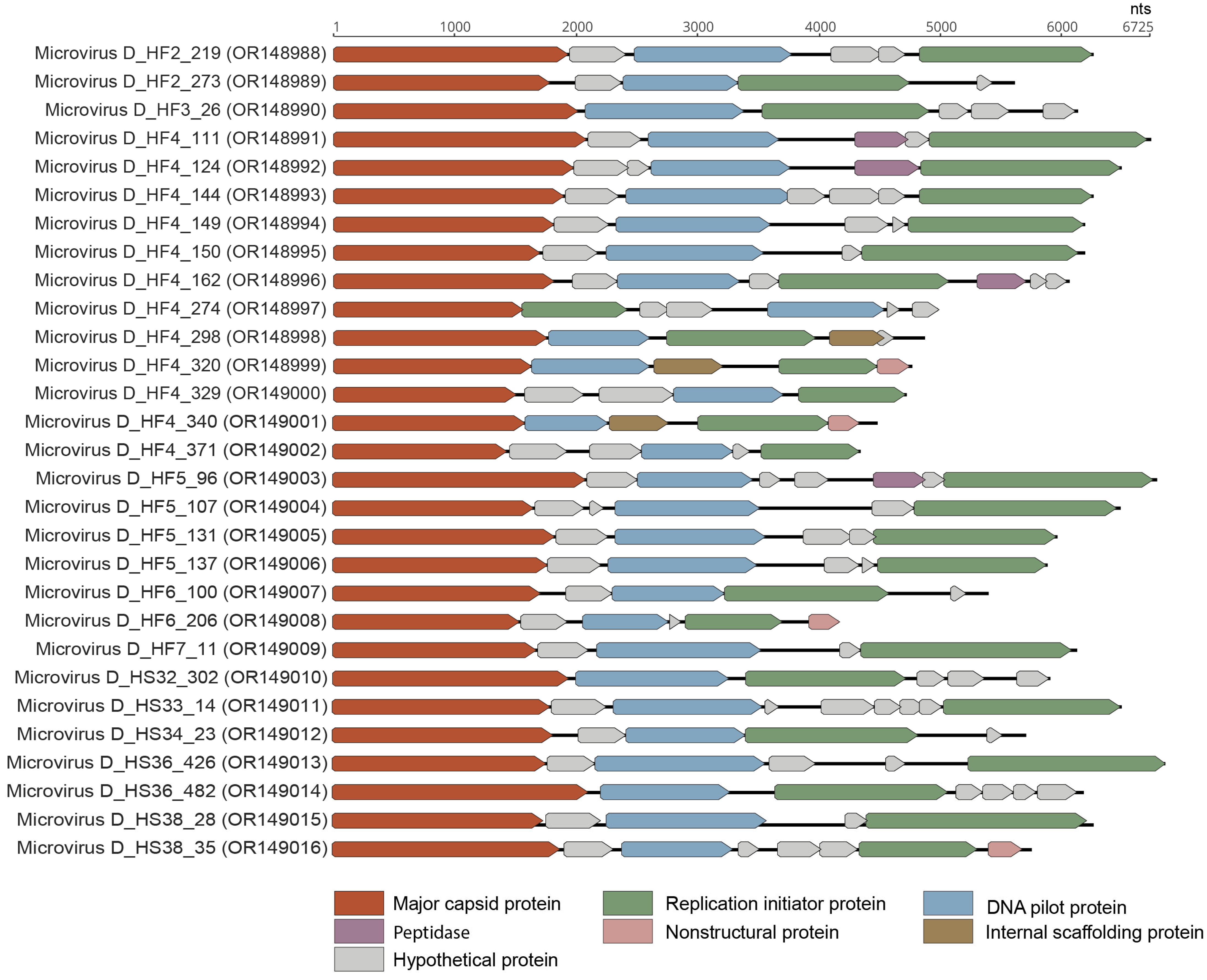
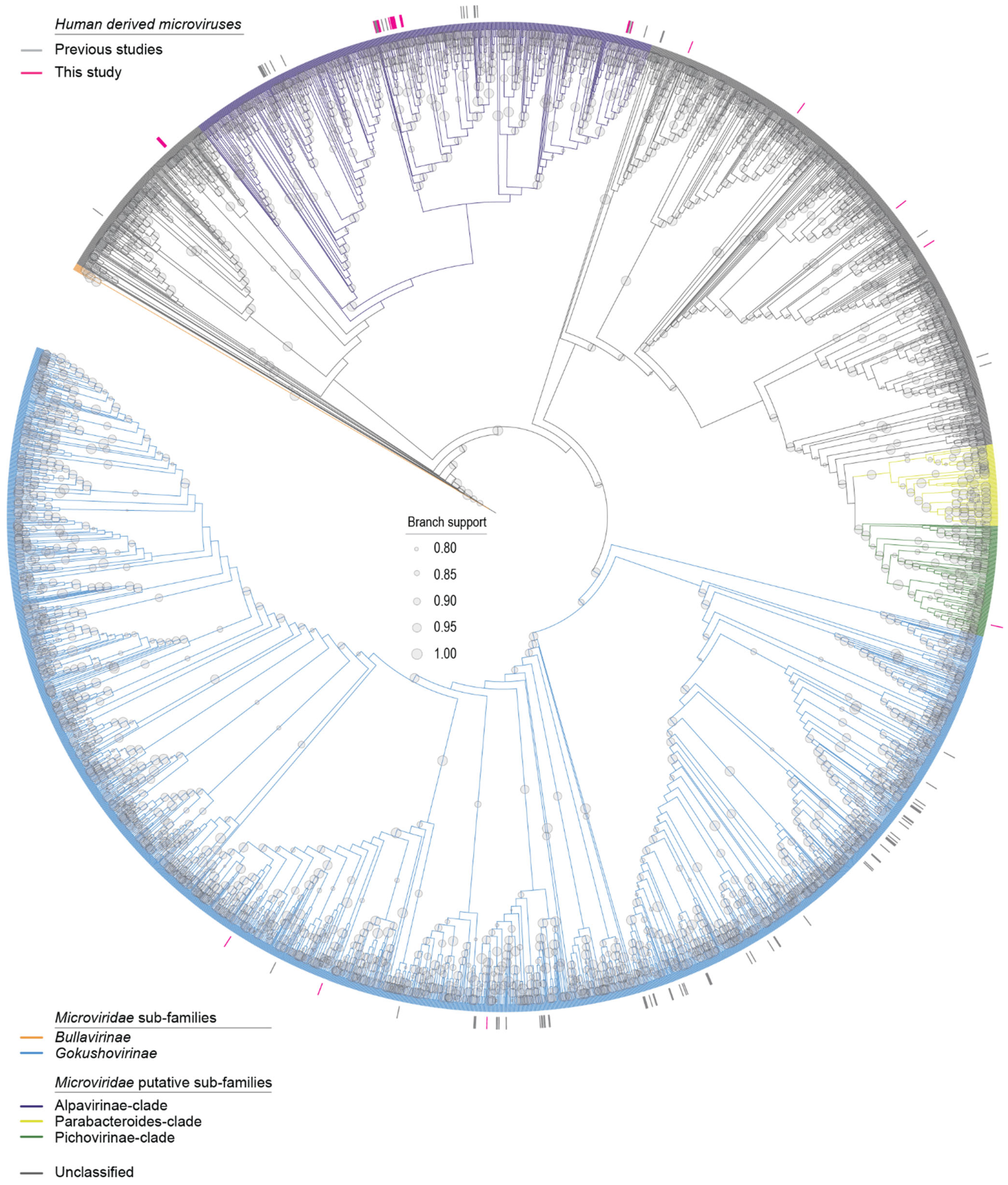
3.4. Microviruses
3.5. Inoviruses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liang, G.; Bushman, F.D. The human virome: Assembly, composition and host interactions. Nat. Rev. Genet. 2021, 19, 514–527. [Google Scholar] [CrossRef]
- Shaiber, A.; Willis, A.D.; Delmont, T.O.; Roux, S.; Chen, L.-X.; Schmid, A.C.; Yousef, M.; Watson, A.R.; Lolans, K.; Esen, Ö.C.; et al. Functional and genetic markers of niche partitioning among enigmatic members of the human oral microbiome. Genome Biol. 2020, 21, 1–35. [Google Scholar] [CrossRef]
- Li, S.; Guo, R.; Zhang, Y.; Li, P.; Chen, F.; Wang, X.; Li, J.; Jie, Z.; Lv, Q.; Jin, H.; et al. A catalog of 48,425 nonredundant viruses from oral metagenomes expands the horizon of the human oral virome. iScience 2022, 25, 104418. [Google Scholar] [CrossRef]
- Ho, S.X.; Min, N.; Wong, E.P.Y.; Chong, C.Y.; Chu, J.J.H. Characterization of oral virome and microbiome revealed distinctive microbiome disruptions in paediatric patients with hand, foot and mouth disease. npj Biofilms Microbiomes 2021, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Abeles, S.R.; Ly, M.; Santiago-Rodriguez, T.M.; Pride, D.T. Effects of Long Term Antibiotic Therapy on Human Oral and Fecal Viromes. PLoS ONE 2015, 10, e0134941. [Google Scholar] [CrossRef]
- Kinsella, C.M.; Deijs, M.; Becker, C.; Broekhuizen, P.; van Gool, T.; Bart, A.; Schaefer, A.S.; van der Hoek, L. Host prediction for disease-associated gastrointestinal cressdnaviruses. Virus Evol. 2022, 8, veac087. [Google Scholar] [CrossRef]
- Guidry, J.T.; Birdwell, C.E.; Scott, R.S. Epstein-Barr virus in the pathogenesis of oral cancers. Oral Dis. 2018, 24, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Arze, C.A.; Springer, S.; Dudas, G.; Patel, S.; Bhattacharyya, A.; Swaminathan, H.; Brugnara, C.; Delagrave, S.; Ong, T.; Kahvejian, A.; et al. Global genome analysis reveals a vast and dynamic anellovirus landscape within the human virome. Cell Host Microbe 2021, 29, 1305–1315.e6. [Google Scholar] [CrossRef] [PubMed]
- Abeles, S.R.; Robles-Sikisaka, R.; Ly, M.; Lum, A.G.; Salzman, J.; Boehm, T.K.; Pride, D.T. Human oral viruses are personal, persistent and gender-consistent. ISME J. 2014, 8, 1753–1767. [Google Scholar] [CrossRef]
- Tisza, M.J.; Buck, C.B. A catalog of tens of thousands of viruses from human metagenomes reveals hidden associations with chronic diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2023202118. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.; Skinner, M.L.; Woelfel, T.; Carpenter, T.; Haggerty, K.P. Implementing Self-Collection of Biological Specimens with a Diverse Sample. Field Methods 2012, 25, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.P.; Koh, D.; Choo, S.G.; Ng, V.; Fu, Q. Effect of storage conditions on the extraction of PCR-quality genomic DNA from saliva. Clin. Chim. Acta 2004, 343, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Oskis, A.; Loveday, C.; Hucklebridge, F.; Thorn, L.; Clow, A. Diurnal patterns of salivary cortisol across the adolescent period in healthy females. Psychoneuroendocrinology 2009, 34, 307–316. [Google Scholar] [CrossRef]
- Robles-Sikisaka, R.; Ly, M.; Boehm, T.; Naidu, M.; Salzman, J.; Pride, D.T. Association between living environment and human oral viral ecology. ISME J. 2013, 7, 1710–1724. [Google Scholar] [CrossRef] [PubMed]
- Willner, D.; Furlan, M.; Schmieder, R.; Grasis, J.A.; Pride, D.T.; Relman, D.A.; Angly, F.E.; McDole, T.; Mariella, R.P.; Rohwer, F.; et al. Metagenomic detection of phage-encoded platelet-binding factors in the human oral cavity. Proc. Natl. Acad. Sci. USA 2011, 108, 4547–4553. [Google Scholar] [CrossRef] [PubMed]
- DiGiulio, D.B.; Callahan, B.J.; McMurdie, P.J.; Costello, E.K.; Lyell, D.J.; Robaczewska, A.; Sun, C.L.; Goltsman, D.S.A.; Wong, R.J.; Shaw, G.; et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc. Natl. Acad. Sci. USA 2015, 112, 11060–11065. [Google Scholar] [CrossRef]
- Goltsman, D.S.A.; Sun, C.L.; Proctor, D.M.; DiGiulio, D.B.; Robaczewska, A.; Thomas, B.C.; Shaw, G.M.; Stevenson, D.K.; Holmes, S.P.; Banfield, J.F.; et al. Metagenomic analysis with strain-level resolution reveals fine-scale variation in the human pregnancy microbiome. Genome Res. 2018, 28, 1467–1480. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.J.; Belford, A.K.; Domínguez-Huerta, G.; Bolduc, B.; Buck, C.B. Cenote-Taker 2 democratizes virus discovery and sequence annotation. Virus Evol. 2021, 7, veaa100. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap Short-Read Aligner, and Other Bioinformatics Tools. 2015. Available online: http://sourceforge.net/projects/bbmap/ (accessed on 15 August 2016).
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Stöver, B.C.; Müller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, D.; Varsani, A.; Krupovic, M. Pervasive Chimerism in the Replication-Associated Proteins of Uncultured Single-Stranded DNA Viruses. Viruses 2018, 10, 187. [Google Scholar] [CrossRef]
- Zallot, R.; Oberg, N.; Gerlt, J.A. The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 2019, 58, 4169–4182. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chrzastek, K.; Kraberger, S.; Schmidlin, K.; Fontenele, R.S.; Kulkarni, A.; Chappell, L.; Dufour-Zavala, L.; Kapczynski, D.R.; Varsani, A. Diverse Single-Stranded DNA Viruses Identified in Chicken Buccal Swabs. Microorganisms 2021, 9, 2602. [Google Scholar] [CrossRef] [PubMed]
- Custer, J.M.; White, R.; Taylor, H.; Schmidlin, K.; Fontenele, R.S.; Stainton, D.; Kraberger, S.; Briskie, J.V.; Varsani, A. Diverse single-stranded DNA viruses identified in New Zealand (Aotearoa) South Island robin (Petroica australis) fecal samples. Virology 2021, 565, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Fontenele, R.S.; Lacorte, C.; Lamas, N.S.; Schmidlin, K.; Varsani, A.; Ribeiro, S.G. Single Stranded DNA Viruses Associated with Capybara Faeces Sampled in Brazil. Viruses 2019, 11, 710. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.; Larsen, B.B.; Otto, H.W.; Potticary, A.L.; Kraberger, S.; Custer, J.M.; Suazo, C.; Upham, N.S.; Worobey, M.; Van Doorslaer, K.; et al. Diverse DNA virus genomes identified in fecal samples of Mexican free-tailed bats (Tadarida brasiliensis) captured in Chiricahua Mountains of southeast Arizona (USA). Virology 2023, 580, 98–111. [Google Scholar] [CrossRef]
- Kraberger, S.; Schmidlin, K.; Fontenele, R.S.; Walters, M.; Varsani, A. Unravelling the Single-Stranded DNA Virome of the New Zealand Blackfly. Viruses 2019, 11, 532. [Google Scholar] [CrossRef]
- Levy, H.; Fontenele, R.S.; Harding, C.; Suazo, C.; Kraberger, S.; Schmidlin, K.; Djurhuus, A.; Black, C.E.; Hart, T.; Smith, A.L.; et al. Identification and Distribution of Novel Cressdnaviruses and Circular Molecules in Four Penguin Species in South Georgia and the Antarctic Peninsula. Viruses 2020, 12, 1029. [Google Scholar] [CrossRef]
- Lund, M.C.; Larsen, B.B.; Rowsey, D.M.; Otto, H.W.; Gryseels, S.; Kraberger, S.; Custer, J.M.; Steger, L.; Yule, K.M.; Harris, R.E.; et al. Using archived and biocollection samples towards deciphering the DNA virus diversity associated with rodent species in the families cricetidae and heteromyidae. Virology 2023, 585, 42–60. [Google Scholar] [CrossRef]
- Orton, J.P.; Morales, M.; Fontenele, R.S.; Schmidlin, K.; Kraberger, S.; Leavitt, D.J.; Webster, T.H.; Wilson, M.A.; Kusumi, K.; Dolby, G.A.; et al. Virus Discovery in Desert Tortoise Fecal Samples: Novel Circular Single-Stranded DNA Viruses. Viruses 2020, 12, 143. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gascuel, O. Approximate Likelihood-Ratio Test for Branches: A Fast, Accurate, and Powerful Alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2020, 39, 578–585. [Google Scholar] [CrossRef]
- Kaczorowska, J.; Deijs, M.; Klein, M.; Bakker, M.; Jebbink, M.F.; Sparreboom, M.; Kinsella, C.M.; Timmerman, A.L.; van der Hoek, L. Diversity and Long-Term Dynamics of Human Blood Anelloviruses. J. Virol. 2022, 96, e0010922. [Google Scholar] [CrossRef] [PubMed]
- Kraberger, S.; Opriessnig, T.; Celer, V.; Maggi, F.; Okamoto, H.; Blomström, A.-L.; Cadar, D.; Harrach, B.; Biagini, P.; Varsani, A. Taxonomic updates for the genus Gyrovirus (family Anelloviridae): Recognition of several new members and establishment of species demarcation criteria. Arch. Virol. 2021, 166, 2937–2942. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Opriessnig, T.; Celer, V.; Maggi, F.; Okamoto, H.; Blomström, A.-L.; Cadar, D.; Harrach, B.; Biagini, P.; Kraberger, S. Taxonomic update for mammalian anelloviruses (family Anelloviridae). Arch. Virol. 2021, 166, 2943–2953. [Google Scholar] [CrossRef]
- Okamoto, H. TT Viruses in Animals; Springer: Berlin/Heidelberg, Germany, 2009; pp. 35–52. [Google Scholar]
- Bigarré, L.; Beven, V.; de Boisséson, C.; Grasland, B.; Rose, N.; Biagini, P.; Jestin, A. Pig anelloviruses are highly prevalent in swine herds in France. J. Gen. Virol. 2005, 86, 631–635. [Google Scholar] [CrossRef]
- Collins, C.L.; Kraberger, S.; Fontenele, R.S.; Faleye, T.O.C.; Adams, D.; Adhikari, S.; Sandrolini, H.; Finnerty, S.; Halden, R.U.; Scotch, M.; et al. Genome Sequences of Anelloviruses, Genomovirus, and Papillomavirus Isolated from Nasal Pharyngeal Swabs. Genome Announc. 2022, 11, e0068122. [Google Scholar] [CrossRef]
- Spandole, S.; Cimponeriu, D.; Berca, L.M.; Mihăescu, G. Human anelloviruses: An update of molecular, epidemiological and clinical aspects. Arch. Virol. 2015, 160, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Amatya, R.; Deem, S.L.; Porton, I.J.; Wang, D.; Lim, E.S. Complete Genome Sequence of Torque teno indri virus 1, a Novel Anellovirus in Blood from a Free-Living Lemur. Genome Announc. 2017, 5, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Butkovic, A.; Kraberger, S.; Smeele, Z.; Martin, D.P.; Schmidlin, K.; Fontenele, R.S.; Shero, M.R.; Beltran, R.S.; Kirkham, A.L.; Aleamotu’a, M.; et al. Evolution of anelloviruses from a circovirus-like ancestor through gradual augmentation of the jelly-roll capsid protein. Virus Evol. 2023, 9, vead035. [Google Scholar] [CrossRef]
- Cordey, S.; Laubscher, F.; Hartley, M.-A.; Junier, T.; Keitel, K.; Docquier, M.; Guex, N.; Iseli, C.; Vieille, G.; Le Mercier, P.; et al. Blood virosphere in febrile Tanzanian children. Emerg. Microbes Infect. 2021, 10, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Varsani, A. A 2021 taxonomy update for the family Smacoviridae. Arch. Virol. 2021, 166, 3245–3253. [Google Scholar] [CrossRef]
- Krupovic, M.; Varsani, A.; Kazlauskas, D.; Breitbart, M.; Delwart, E.; Rosario, K.; Yutin, N.; Wolf, Y.I.; Harrach, B.; Zerbini, F.M.; et al. Cressdnaviricota: A Virus Phylum Unifying Seven Families of Rep-Encoding Viruses with Single-Stranded, Circular DNA Genomes. J. Virol. 2020, 94, e00582-20. [Google Scholar] [CrossRef]
- Krupovic, M.; Varsani, A. Naryaviridae, Nenyaviridae, and Vilyaviridae: Three new families of single-stranded DNA viruses in the phylum Cressdnaviricota. Arch. Virol. 2022, 167, 2907–2921. [Google Scholar] [CrossRef]
- Noell, K.; Kolls, J.K. Further Defining the Human Virome using NGS: Identification of Redondoviridae. Cell Host Microbe 2019, 25, 634–635. [Google Scholar] [CrossRef]
- Abbas, A.A.; Taylor, L.J.; Dothard, M.I.; Leiby, J.S.; Fitzgerald, A.S.; Khatib, L.A.; Collman, R.G.; Bushman, F.D. Redondoviridae, a Family of Small, Circular DNA Viruses of the Human Oro-Respiratory Tract Associated with Periodontitis and Critical Illness. Cell Host Microbe 2019, 25, 719–729.e4. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.J.; Dothard, M.I.; Rubel, M.A.; Allen, A.A.; Hwang, Y.; Roche, A.M.; Graham-Wooten, J.; Fitzgerald, A.S.; Khatib, L.A.; Ranciaro, A.; et al. Redondovirus Diversity and Evolution on Global, Individual, and Molecular Scales. J. Virol. 2021, 95, e0081721. [Google Scholar] [CrossRef]
- Keeler, E.L.; Merenstein, C.; Reddy, S.; Taylor, L.J.; Cobián-Güemes, A.G.; Zankharia, U.; Collman, R.G.; Bushman, F.D. Widespread, human-associated redondoviruses infect the commensal protozoan Entamoeba gingivalis. Cell Host Microbe 2023, 31, 58–68.e5. [Google Scholar] [CrossRef] [PubMed]
- Male, M.F.; Kraberger, S.; Stainton, D.; Kami, V.; Varsani, A. Cycloviruses, gemycircularviruses and other novel replication-associated protein encoding circular viruses in Pacific flying fox (Pteropus tonganus) faeces. Infect. Genet. Evol. 2016, 39, 279–292. [Google Scholar] [CrossRef]
- Pearson, V.M.; Caudle, S.B.; Rokyta, D.R. Viral recombination blurs taxonomic lines: Examination of single-stranded DNA viruses in a wastewater treatment plant. PeerJ 2016, 4, e2585. [Google Scholar] [CrossRef]
- Trubl, G.; Roux, S.; Borton, M.A.; Varsani, A.; Li, Y.F.; Sun, C.; Jang, H.B.; Woodcroft, B.; Tyson, G.; Wrighton, K.; et al. Population ecology and potential biogeochemical impacts of ssDNA and dsDNA soil viruses along a permafrost thaw gradient. bioRxiv 2023, 2023-06. [Google Scholar]
- Pride, D.T.; Salzman, J.; Haynes, M.; Rohwer, F.; Davis-Long, C.; White, R.A.; Loomer, P.; Armitage, G.C.; Relman, D.A. Evidence of a robust resident bacteriophage population revealed through analysis of the human salivary virome. ISME J. 2011, 6, 915–926. [Google Scholar] [CrossRef]
- Zablocki, O.; van Zyl, L.; Adriaenssens, E.M.; Rubagotti, E.; Tuffin, M.; Cary, S.C.; Cowan, D. High-Level Diversity of Tailed Phages, Eukaryote-Associated Viruses, and Virophage-Like Elements in the Metaviromes of Antarctic Soils. Appl. Environ. Microbiol. 2014, 80, 6888–6897. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M. Phage Diversity in the Human Gut Microbiome: A Taxonomist’s Perspective. mSystems 2021, 6, e0079921. [Google Scholar] [CrossRef]
- Happel, A.-U.; Varsani, A.; Balle, C.; Passmore, J.-A.; Jaspan, H. The Vaginal Virome—Balancing Female Genital Tract Bacteriome, Mucosal Immunity, and Sexual and Reproductive Health Outcomes? Viruses 2020, 12, 832. [Google Scholar] [CrossRef]
- Benler, S.; Yutin, N.; Antipov, D.; Rayko, M.; Shmakov, S.; Gussow, A.B.; Pevzner, P.; Koonin, E.V. Thousands of previously unknown phages discovered in whole-community human gut metagenomes. Microbiome 2021, 9, 1–17. [Google Scholar] [CrossRef]
- González, B.; Monroe, L.; Li, K.; Yan, R.; Wright, E.; Walter, T.; Kihara, D.; Weintraub, S.T.; Thomas, J.A.; Serwer, P.; et al. Phage G Structure at 6.1 Å Resolution, Condensed DNA, and Host Identity Revision to a Lysinibacillus. J. Mol. Biol. 2020, 432, 4139–4153. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Sommers, P.; Chatterjee, A.; Varsani, A.; Trubl, G. Integrating Viral Metagenomics into an Ecological Framework. Annu. Rev. Virol. 2021, 8, 133–158. [Google Scholar] [CrossRef]
- Benaud, N.; Chelliah, D.S.; Wong, S.Y.; Ferrari, B.C. Soil substrate culturing approaches recover diverse members of Actinomycetota from desert soils of Herring Island, East Antarctica. Extremophiles 2022, 26, 24. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Zhou, Y.; Wang, C.; Wu, B.; Wan, J. Actinomyces and Alimentary Tract Diseases: A Review of Its Biological Functions and Pathology. BioMed Res. Int. 2018, 2018, 3820215. [Google Scholar]
- Panwar, D.; Shubhashini, A.; Kapoor, M. Complex alpha and beta mannan foraging by the human gut bacteria. Biotechnol. Adv. 2023, 66, 108166. [Google Scholar] [CrossRef]
- Lupu, V.V.; Raileanu, A.A.; Mihai, C.M.; Morariu, I.D.; Lupu, A.; Starcea, I.M.; Frasinariu, O.E.; Mocanu, A.; Dragan, F.; Fotea, S. The Implication of the Gut Microbiome in Heart Failure. Cells 2023, 12, 1158. [Google Scholar] [CrossRef]
- Collins, C.L.; DeNardo, D.F.; Blake, M.; Norton, J.; Schmidlin, K.; Fontenele, R.S.; Wilson, M.A.; Kraberger, S.; Varsani, A. Genome Sequences of Microviruses Identified in Gila Monster Feces. Genome Announc. 2021, 10, e00163-21. [Google Scholar] [CrossRef]
- Kirchberger, P.C.; Martinez, Z.A.; Ochman, H. Organizing the Global Diversity of Microviruses. mBio 2022, 13, e0058822. [Google Scholar] [CrossRef] [PubMed]
- Cherwa, J.E.; Fane, B.A. Microviridae: Microviruses and Gokushoviruses. eLS 2011, 1–10. [Google Scholar] [CrossRef]
- Uchiyama, A.; Chen, M.; Fane, B.A. Characterization and Function of Putative Substrate Specificity Domain in Microvirus External Scaffolding Proteins. J. Virol. 2007, 81, 8587–8592. [Google Scholar] [CrossRef] [PubMed]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.M. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef]
- Roux, S.; Krupovic, M.; Daly, R.A.; Borges, A.L.; Nayfach, S.; Schulz, F.; Sharrar, A.; Carnevali, P.B.M.; Cheng, J.-F.; Ivanova, N.N.; et al. Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth’s biomes. Nat. Microbiol. 2019, 4, 1895–1906. [Google Scholar] [CrossRef]
- Knezevic, P.; Adriaenssens, E.M. ICTV Report Consortium ICTV Virus Taxonomy Profile: Inoviridae. J. Gen. Virol. 2021, 102, 001614. [Google Scholar] [CrossRef]
- Ilyina, T.S. Filamentous bacteriophages and their role in the virulence and evolution of pathogenic bacteria. Mol. Genet. Microbiol. Virol. 2015, 30, 1–9. [Google Scholar] [CrossRef]
- Loh, B.; Haase, M.; Mueller, L.; Kuhn, A.; Leptihn, S. The Transmembrane Morphogenesis Protein gp1 of Filamentous Phages Contains Walker A and Walker B Motifs Essential for Phage Assembly. Viruses 2017, 9, 73. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, M.; Falkow, S.; Rosenberg, E.; Schleifer, K.-H.; Stackebrandt, E. The Prokaryotes: Volume 5: Proteobacteria: Alpha and Beta Subclasses; Springer: Berlin/Heidelberg, Germany, 2006; pp. 602–647. [Google Scholar]
- Thompson, S.A.; Wang, L.L.; West, A.; Sparling, P.F. Neisseria meningitidis produces iron-regulated proteins related to the RTX family of exoproteins. J. Bacteriol. 1993, 175, 811–818. [Google Scholar] [CrossRef]
- Gholizadeh, P.; Pormohammad, A.; Eslami, H.; Shokouhi, B.; Fakhrzadeh, V.; Kafil, H.S. Oral pathogenesis of Aggregatibacter actinomycetemcomitans. Microb. Pathog. 2017, 113, 303–311. [Google Scholar] [CrossRef]
- Corbel, M.J. 35—Yersinia, Pasteurella and Francisella: Plague; Pseudotuberculosis; Mesenteric Adenitis; Pasteurellosis; Tularaemia. In Medical Microbiology, 18th ed.; Greenwood, D., Barer, M., Slack, R., Irving, W., Eds.; Churchill Livingstone: London, UK, 2012; pp. 350–358. [Google Scholar]
- Aulik, N.A.; Hellenbrand, K.M.; Klos, H.; Czuprynski, C.J. Mannheimia haemolytica and its leukotoxin cause neutrophil extracellular trap formation by bovine neutrophils. Infect. Immun. 2010, 78, 4454–4466. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | Class | Family | Number of Virus Genomes Identified | GenBank Accession Number |
|---|---|---|---|---|
| N/A | N/A | Anelloviridae | 3 | OR148953, OR148954, OR148955 |
| Cressdnaviricota | Arfiviricetes | Redondoviridae | 5 | OR148956, OR148957, OR148962, OR148963, OR148964 |
| N/A | Unclassified | 4 | OR148958, OR148959, OR148960, OR148961 | |
| Uroviricota | Caudoviricetes | N/A | 4 | OR148984, OR148985, OR148986, OR148987 |
| Phixviricota | Malgrandaviricetes | Microviridae | 29 | OR148988, OR148989, OR148990, OR148991, OR148992, OR148993, OR148994, OR148995, OR148996, OR148997, OR148998, OR148999, OR149000, OR149001, OR149002, OR149003, OR149004, OR149005, OR149006, OR149007, OR149008, OR149009, OR149010, OR149011, OR149012, OR149013, OR149014, OR149015, OR149016 |
| Hofneiviricota | Faserviricetes | Inoviridae | 19 | OR148965, OR148966, OR148967, OR148968, OR148969, OR148970, OR148971, OR148972, OR148973, OR148974, OR148975, OR148976, OR148977, OR148978, OR148979, OR148980, OR148981, OR148982, OR148983 |
| Accession Number | Genome Length (nt) | Data for Top BLASTn Hit | |||
|---|---|---|---|---|---|
| Best Hit (NCBI Accession No.) | Percent Identity (%) | Query Cover (%) | Microvirus Source | ||
| OR148988 | 6252 | BK022849 | 80 | 89 | Human metagenome, USA |
| OR148989 | 5600 | BK050880 | 93 | 91 | Human metagenome, USA |
| OR148990 | 6125 | BK047429 | 74 | 6 | Human metagenome, USA |
| OR148991 | 6725 | BK053955 | 83 | 62 | Human metagenome, USA |
| OR148992 | 6482 | BK040340 | 92 | 100 | Human metagenome, USA |
| OR148993 | 6250 | BK022849 | 78 | 74 | Human metagenome, USA |
| OR148994 | 6185 | BK044794 | 93 | 91 | Human metagenome, USA |
| OR148995 | 6178 | BK051052 | 99 | 100 | Human metagenome, USA |
| OR148996 | 6058 | BK031150 | 73 | 33 | Human metagenome, USA |
| OR148997 | 4966 | MH572463 | 72 | 21 | Ciona robusta intestinal tract, USA |
| OR148998 | 4861 | MT310328 | 71 | 39 | Wastewater metagenome, USA |
| OR148999 | 4753 | MH617740 | 72 | 96 | Minnow tissue metagenome. USA |
| OR149000 | 4708 | MH617589 | 71 | 19 | Minnow tissue metagenome, USA |
| OR149001 | 4645 | MT204053 | 70 | 18 | Freshwater metagenome, Svalbard |
| OR149002 | 4499 | MH617140 | 76 | 38 | Minnow tissue metagenome, USA |
| OR149003 | 7033 | BK041046 | 72 | 53 | Human metagenome, USA |
| OR149004 | 6719 | BK015704 | 94 | 100 | Human metagenome, USA |
| OR149005 | 6177 | BK022849 | 78 | 80 | Human metagenome, USA |
| OR149006 | 6102 | BK044794 | 89 | 91 | Human metagenome, USA |
| OR149007 | 5590 | BK050880 | 92 | 91 | Human metagenome, USA |
| OR149008 | 4311 | BK039346 | 75 | 84 | Human metagenome, USA |
| OR149009 | 6346 | BK039742 | 91 | 100 | Human metagenome, USA |
| OR149010 | 6125 | BK047429 | 74 | 6 | Human metagenome, USA |
| OR149011 | 6378 | BK042793 | 97 | 93 | Human metagenome, USA |
| OR149012 | 5601 | BK050880 | 93 | 91 | Human metagenome, USA |
| OR149013 | 6729 | BK049539 | 73 | 95 | Human metagenome, USA |
| OR149014 | 6067 | MG883710 | 84 | 92 | Human nasopharyngeal cavity, China |
| OR149015 | 6152 | BK053938 | 90 | 90 | Human metagenome, USA |
| OR149016 | 5654 | MK765647 | 70 | 3 | Tortoise feces, USA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paietta, E.N.; Kraberger, S.; Custer, J.M.; Vargas, K.L.; Espy, C.; Ehmke, E.; Yoder, A.D.; Varsani, A. Characterization of Diverse Anelloviruses, Cressdnaviruses, and Bacteriophages in the Human Oral DNA Virome from North Carolina (USA). Viruses 2023, 15, 1821. https://doi.org/10.3390/v15091821
Paietta EN, Kraberger S, Custer JM, Vargas KL, Espy C, Ehmke E, Yoder AD, Varsani A. Characterization of Diverse Anelloviruses, Cressdnaviruses, and Bacteriophages in the Human Oral DNA Virome from North Carolina (USA). Viruses. 2023; 15(9):1821. https://doi.org/10.3390/v15091821
Chicago/Turabian StylePaietta, Elise N., Simona Kraberger, Joy M. Custer, Karla L. Vargas, Claudia Espy, Erin Ehmke, Anne D. Yoder, and Arvind Varsani. 2023. "Characterization of Diverse Anelloviruses, Cressdnaviruses, and Bacteriophages in the Human Oral DNA Virome from North Carolina (USA)" Viruses 15, no. 9: 1821. https://doi.org/10.3390/v15091821