
Targeting SARS-CoV-2 Macrodomain-1 to Restore the Innate Immune Response Using In Silico Screening of Medicinal Compounds and Free Energy Calculation Approaches

 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Structures, Sequence Retrieval, and Modeling
2.2. Molecular Screening of Medicinal Compound Databases
2.3. Molecular Dynamic Simulation (MD)
2.4. Binding Free Energy Calculations
2.5. Determination of Dissociation Constant and Bioactivity for the Top Hits
3. Results
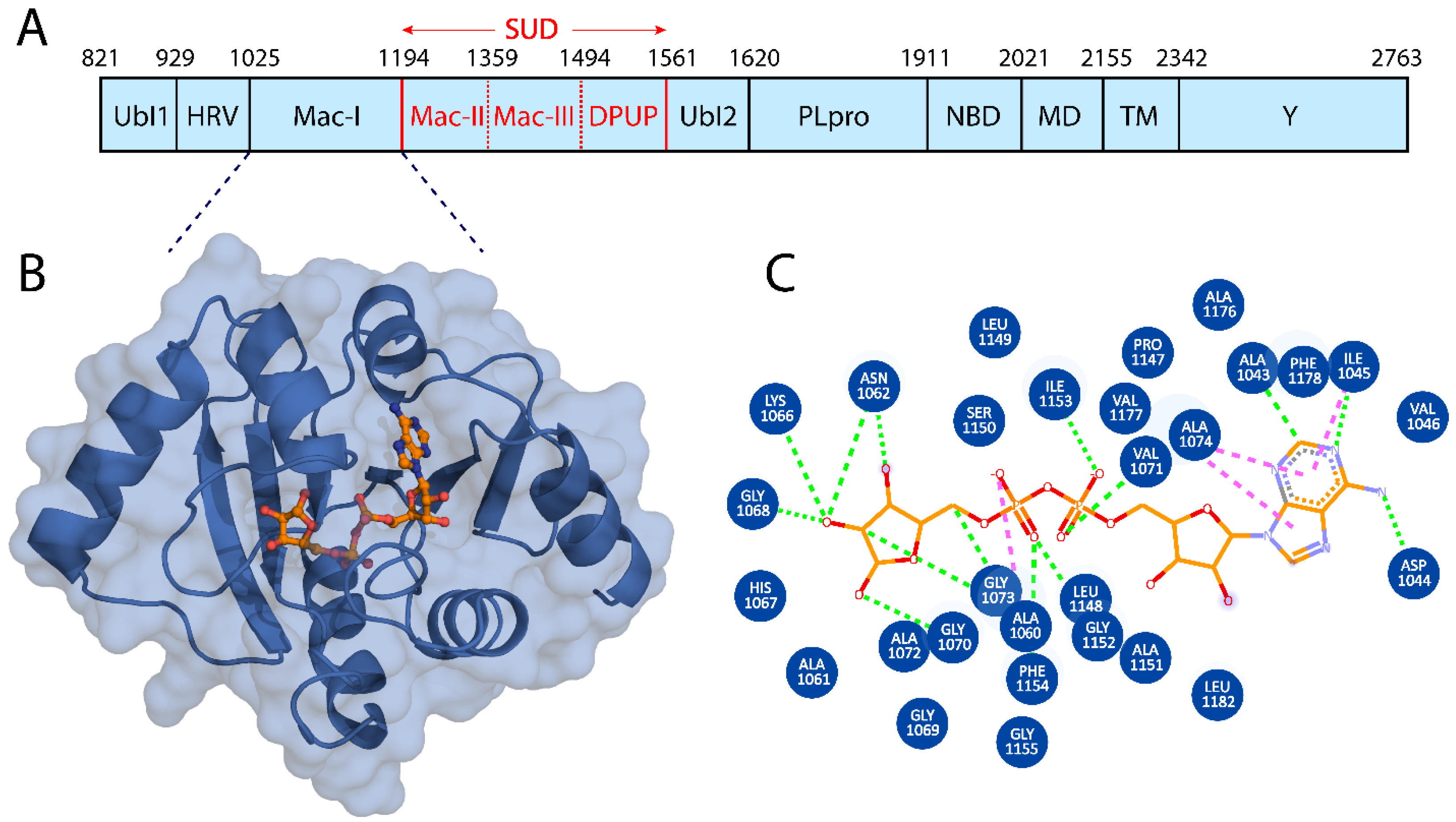
3.1. Macrodomain I Structural Modeling
3.2. Discovery of Small-Molecule Inhibitors by Screening Large Libraries
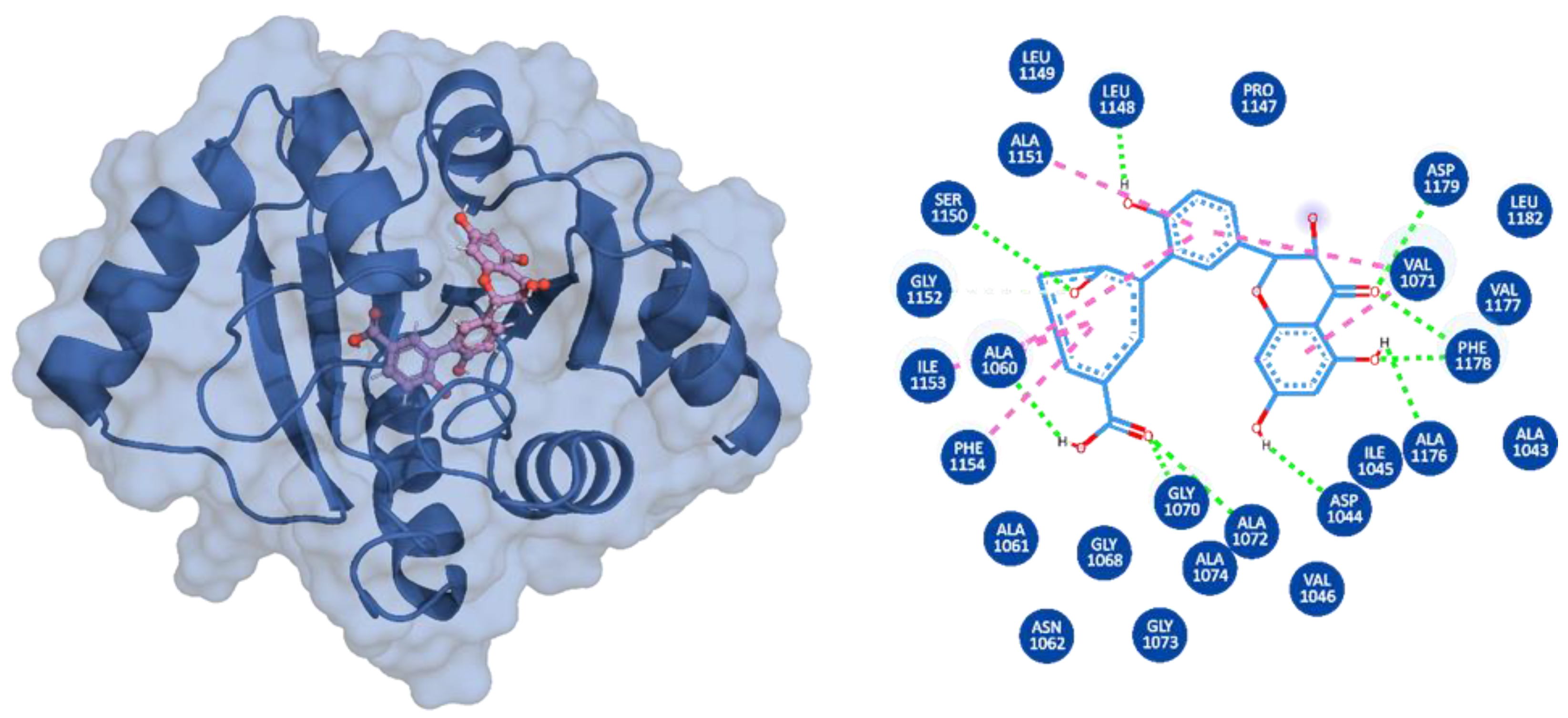
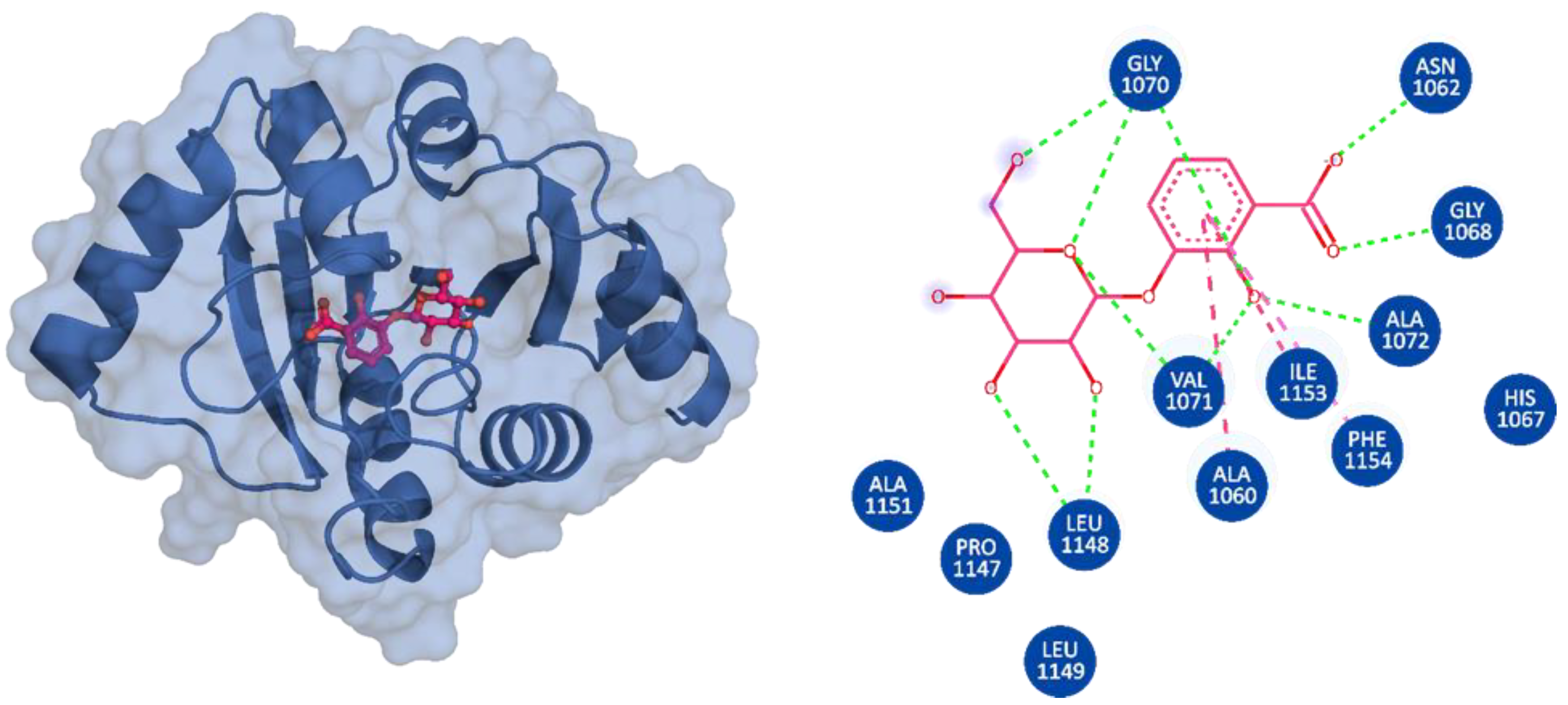
3.3. Binding Modes of the Selected Compounds
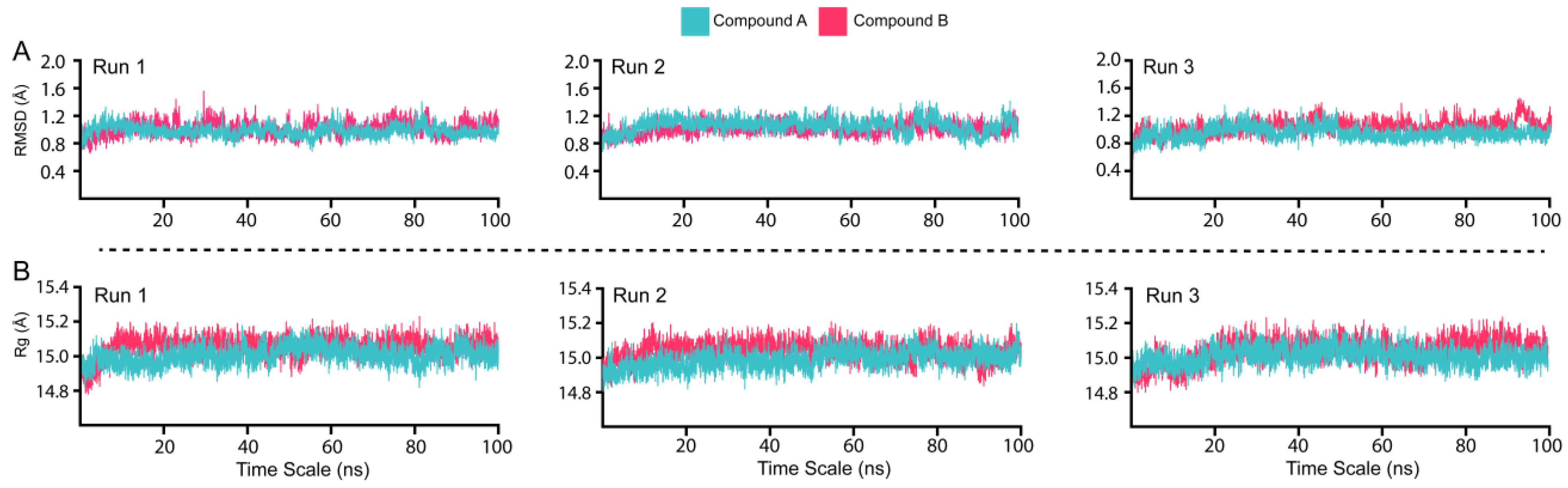
3.4. Dynamic Stability and Compactness Assessment
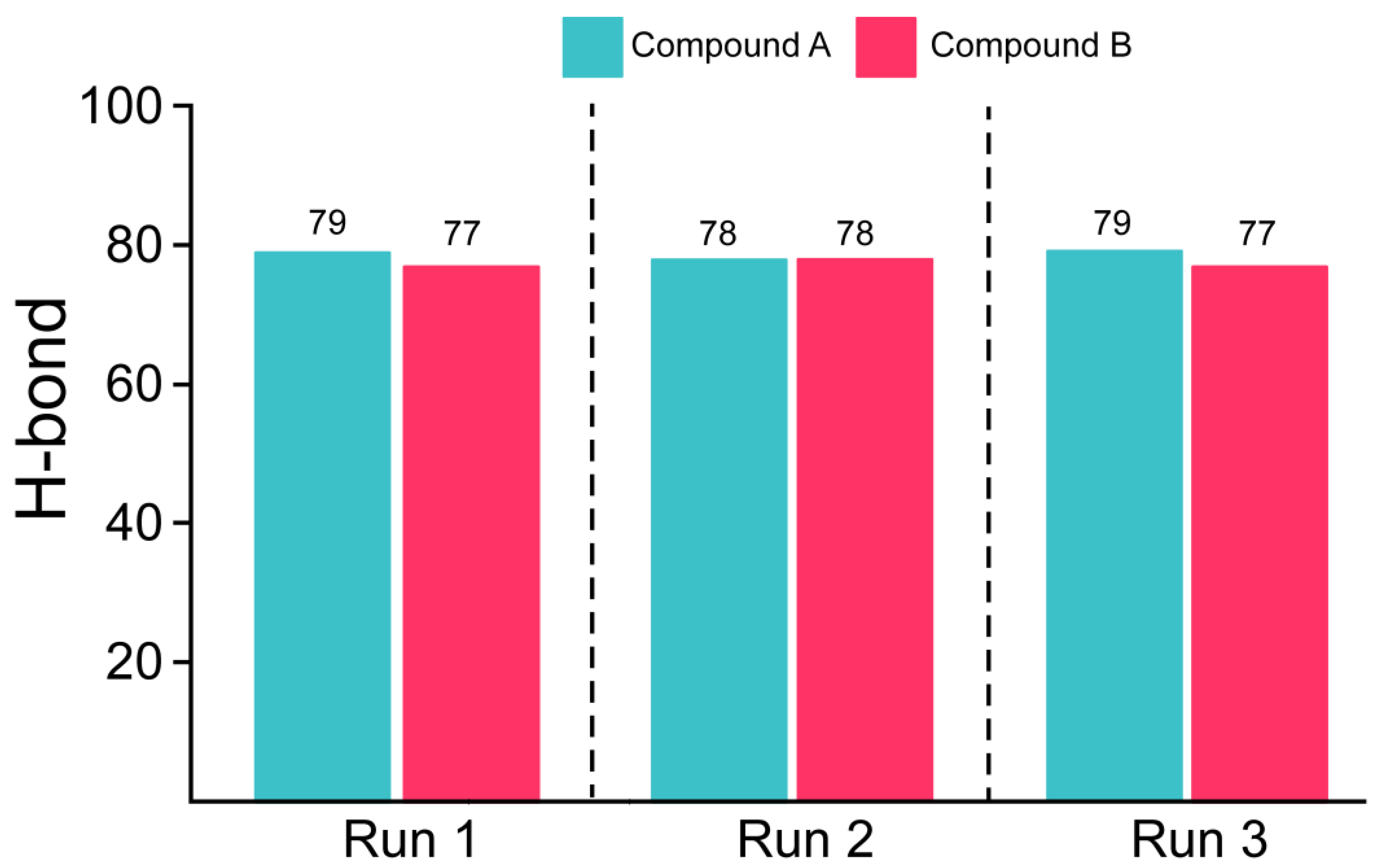
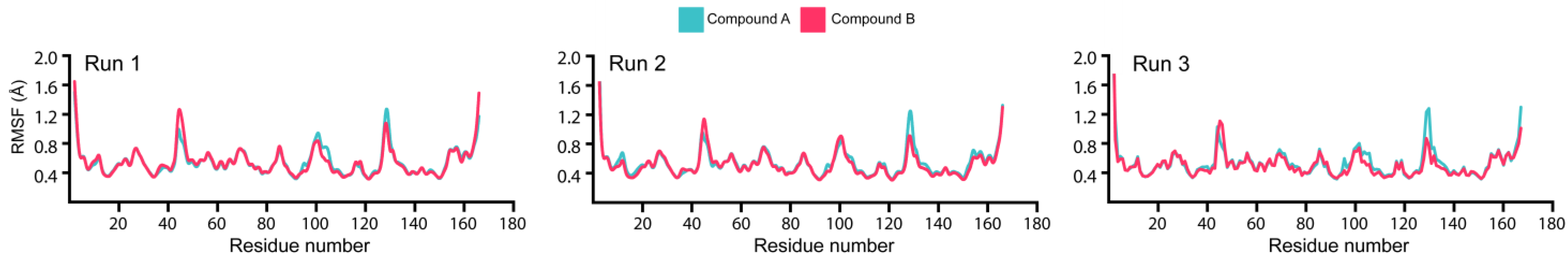
3.5. Estimation of Hydrogen Bonding and Residual Flexibility
3.6. Binding Free Energy Estimation
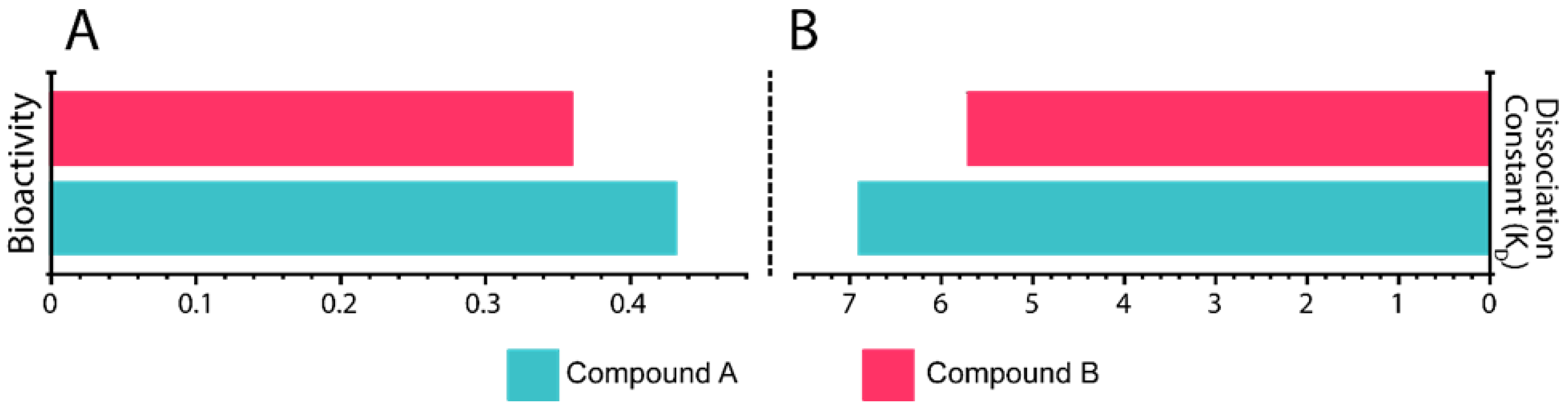
3.7. In Silico Bioactivity and KD Estimation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Masters, P.S. Coronavirus genomic RNA packaging. Virology 2019, 537, 198–207. [Google Scholar] [CrossRef]
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [CrossRef]
- Mohammad, A.; Alshawaf, E.; Marafie, S.K.; Abu-Farha, M.; Abubaker, J.; Al-Mulla, F. Higher binding affinity of Furin to SARS-CoV-2 spike (S) protein D614G could be associated with higher SARS-CoV-2 infectivity. Int. J. Infect. Dis. 2020, 103, 611–616. [Google Scholar] [CrossRef]
- Haddad, D.; John, S.E.; Mohammad, A.; Hammad, M.M.; Hebbar, P.; Channanath, A.; Nizam, R.; Al-Qabandi, S.; Al Madhoun, A.; Alshukry, A.; et al. SARS-CoV-2: Possible recombination and emergence of potentially more virulent strains. PLoS ONE 2021, 16, e0251368. [Google Scholar] [CrossRef]
- Eaaswarkhanth, M.; Madhoun, A.A.; Al-Mulla, F. Could the D614 G substitution in the SARS-CoV-2 spike (S) protein be associated with higher COVID-19 mortality? Int. J. Infect. Dis. 2020, 96, 459–460. [Google Scholar] [CrossRef]
- Yadav, P.D.; Nyayanit, D.A.; Sahay, R.R.; Sarkale, P.; Pethani, J.; Patil, S.; Baradkar, S.; Potdar, V.; Patil, D.Y. Isolation and characterization of the new SARS-CoV-2 variant in travellers from the United Kingdom to India: VUI-202012/01 of the B.1.1.7 lineage. J. Travel Med. 2021, 28, taab009. [Google Scholar] [CrossRef]
- Rambaut, A.; Loman, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.L.; Volz, E. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Genom. Epidemiol. 2020, 1–5. [Google Scholar]
- Mohammad, A.; Abubaker, J.; Al-Mulla, F. Structural modelling of SARS-CoV-2 alpha variant (B.1.1.7) suggests enhanced furin binding and infectivity. Virus Res. 2021, 303, 198522. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.d.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef]
- Funk, T.; Pharris, A.; Spiteri, G.; Bundle, N.; Melidou, A.; Carr, M.; Gonzalez, G.; Garcia-Leon, A.; Crispie, F.; O’Connor, L.; et al. Characteristics of SARS-CoV-2 variants of concern B.1.1.7, B.1.351 or P.1: Data from seven EU/EEA countries, weeks 38/2020 to 10/2021. Euro Surveill. Bull. Eur. Mal. Transm./Eur. Commun. Dis. Bull. 2021, 26, 2100348. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Zhang, L.; Cui, Z.; Li, Q.; Wang, B.; Yu, Y.; Wu, J.; Nie, J.; Ding, R.; Wang, H.; Zhang, Y.; et al. Ten emerging SARS-CoV-2 spike variants exhibit variable infectivity, animal tropism, and antibody neutralization. Commun. Biol. 2021, 4, 1196. [Google Scholar] [CrossRef]
- Yadav, P.D.; Sapkal, G.N.; Abraham, P.; Ella, R.; Deshpande, G.; Patil, D.Y.; Nyayanit, D.A.; Gupta, N.; Sahay, R.R.; Shete, A.M.; et al. Neutralization of Variant under Investigation B.1.617.1 With Sera of BBV152 Vaccinees. Clin. Infect. Dis. 2021, 74, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Krüger, N.; Schulz, S.; Cossmann, A.; Rocha, C.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Moldenhauer, A.S.; Winkler, M.S.; et al. The Omicron variant is highly resistant against antibody-mediated neutralization: Implications for control of the COVID-19 pandemic. Cell 2022, 185, 447–456.e411. [Google Scholar] [CrossRef]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 variant: A new chapter in the COVID-19 pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef]
- Khan, A.; Waris, H.; Rafique, M.; Suleman, M.; Mohammad, A.; Ali, S.S.; Khan, T.; Waheed, Y.; Liao, C.; Wei, D.-Q. The Omicron (B.1.1.529) variant of SARS-CoV-2 binds to the hACE2 receptor more strongly and escapes the antibody response: Insights from structural and simulation data. Int. J. Biol. Macromol. 2022, 200, 438–448. [Google Scholar] [CrossRef] [PubMed]
- CDC. SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html (accessed on 21 December 2022).
- Ghazy, R.M.; Ashmawy, R.; Hamdy, N.A.; Elhadi, Y.A.M.; Reyad, O.A.; Elmalawany, D.; Almaghraby, A.; Shaaban, R.; Taha, S.H.N. Efficacy and Effectiveness of SARS-CoV-2 Vaccines: A Systematic Review and Meta-Analysis. Vaccines 2022, 10, 350. [Google Scholar] [CrossRef]
- Wu, C.-R.; Yin, W.-C.; Jiang, Y.; Xu, H.E. Structure genomics of SARS-CoV-2 and its Omicron variant: Drug design templates for COVID-19. Acta Pharmacol. Sin. 2022, 43, 3021–3033. [Google Scholar] [CrossRef]
- Mohammad, A.; Alshawaf, E.; Marafie, S.K.; Abu-Farha, M.; Al-Mulla, F.; Abubaker, J. Molecular Simulation-Based Investigation of Highly Potent Natural Products to Abrogate Formation of the nsp10–nsp16 Complex of SARS-CoV-2. Biomolecules 2021, 11, 573. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ali, S.S.; Khan, M.T.; Saleem, S.; Ali, A.; Suleman, M.; Babar, Z.; Shafiq, A.; Khan, M.; Wei, D.-Q. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro). J. Biomol. Struct. Dyn. 2021, 39, 4659–4670. [Google Scholar] [CrossRef]
- Chaves, O.A.; Fintelman-Rodrigues, N.; Wang, X.; Sacramento, C.Q.; Temerozo, J.R.; Ferreira, A.C.; Mattos, M.; Pereira-Dutra, F.; Bozza, P.T.; Castro-Faria-Neto, H.C.; et al. Commercially Available Flavonols Are Better SARS-CoV-2 Inhibitors than Isoflavone and Flavones. Viruses 2022, 14, 1458. [Google Scholar] [CrossRef]
- Chaves, O.A.; Lima, C.R.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; de Freitas, C.S.; Vazquez, L.; Temerozo, J.R.; Rocha, M.E.N.; Dias, S.S.G.; Carels, N.; et al. Agathisflavone, a natural biflavonoid that inhibits SARS-CoV-2 replication by targeting its proteases. Int. J. Biol. Macromol. 2022, 222, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Schuller, M.; Correy, G.J.; Gahbauer, S.; Fearon, D.; Wu, T.; Díaz, R.E.; Young, I.D.; Carvalho Martins, L.; Smith, D.H.; Schulze-Gahmen, U.; et al. Fragment binding to the Nsp3 macrodomain of SARS-CoV-2 identified through crystallographic screening and computational docking. Sci. Adv. 2021, 7, eabf8711. [Google Scholar] [CrossRef]
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural biology of SARS-CoV-2: Open the door for novel therapies. Signal Transduct. Target. Ther. 2022, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Pervaiz, N.; Khan, A.; Saleem, S.; Shireen, H.; Wei, D.-Q.; Labrie, V.; Bao, Y.; Abbasi, A.A. Evolutionary and structural analysis of SARS-CoV-2 specific evasion of host immunity. Genes Immun. 2020, 21, 409–419. [Google Scholar] [CrossRef]
- Fehr, A.R.; Jankevicius, G.; Ahel, I.; Perlman, S. Viral Macrodomains: Unique Mediators of Viral Replication and Pathogenesis. Trends Microbiol. 2018, 26, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.; McPherson, R.L.; Griffin, D.E. Macrodomain ADP-ribosylhydrolase and the pathogenesis of infectious diseases. PLoS Pathog. 2018, 14, e1006864. [Google Scholar] [CrossRef]
- Frick, D.N.; Virdi, R.S.; Vuksanovic, N.; Dahal, N.; Silvaggi, N.R. Molecular Basis for ADP-Ribose Binding to the Mac1 Domain of SARS-CoV-2 nsp3. Biochemistry 2020, 59, 2608–2615. [Google Scholar] [CrossRef]
- Alhammad, Y.M.O.; Fehr, A.R. The Viral Macrodomain Counters Host Antiviral ADP-Ribosylation. Viruses 2020, 12, 384. [Google Scholar] [CrossRef]
- Fehr, A.R.; Channappanavar, R.; Jankevicius, G.; Fett, C.; Zhao, J.; Athmer, J.; Meyerholz, D.K.; Ahel, I.; Perlman, S. The Conserved Coronavirus Macrodomain Promotes Virulence and Suppresses the Innate Immune Response during Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 2016, 7, 01721-16. [Google Scholar] [CrossRef]
- Li, C.; Debing, Y.; Jankevicius, G.; Neyts, J.; Ahel, I.; Coutard, B.; Canard, B. Viral Macro Domains Reverse Protein ADP-Ribosylation. J. Virol. 2016, 90, 8478–8486. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Li, X.; Fu, X. The macro domain protein family: Structure, functions, and their potential therapeutic implications. Mutat. Res./Rev. Mutat. Res. 2011, 727, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-H.; Chang, S.-C.; Chiu, Y.-C.; Jiang, B.-C.; Wu, T.-H.; Hsu, C.-H. Structural, biophysical, and biochemical elucidation of the SARS-CoV-2 nonstructural protein 3 macro domain. ACS Infect. Dis. 2020, 6, 2970–2978. [Google Scholar] [CrossRef]
- Srinivasan, S.; Cui, H.; Gao, Z.; Liu, M.; Lu, S.; Mkandawire, W.; Narykov, O.; Sun, M.; Korkin, D. Structural genomics of SARS-CoV-2 indicates evolutionary conserved functional regions of viral proteins. Viruses 2020, 12, 360. [Google Scholar] [CrossRef]
- Hoch, N.C. Host ADP-ribosylation and the SARS-CoV-2 macrodomain. Biochem. Soc. Trans. 2021, 49, 1711–1721. [Google Scholar] [CrossRef]
- Molaei, S.; Dadkhah, M.; Asghariazar, V.; Karami, C.; Safarzadeh, E. The immune response and immune evasion characteristics in SARS-CoV, MERS-CoV, and SARS-CoV-2: Vaccine design strategies. Int. Immunopharmacol. 2021, 92, 107051. [Google Scholar] [CrossRef]
- Claverie, J.M. A Putative Role of de-Mono-ADP-Ribosylation of STAT1 by the SARS-CoV-2 Nsp3 Protein in the Cytokine Storm Syndrome of COVID-19. Viruses 2020, 12, 646. [Google Scholar] [CrossRef]
- Brosey, C.A.; Houl, J.H.; Katsonis, P.; Balapiti-Modarage, L.P.F.; Bommagani, S.; Arvai, A.; Moiani, D.; Bacolla, A.; Link, T.; Warden, L.S.; et al. Targeting SARS-CoV-2 Nsp3 macrodomain structure with insights from human poly(ADP-ribose) glycohydrolase (PARG) structures with inhibitors. Prog. Biophys. Mol. Biol. 2021, 163, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell. Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef]
- Lunić, T.M.; Oalđe, M.M.; Mandić, M.R.; Sabovljević, A.D.; Sabovljević, M.S.; Gašić, U.M.; Duletić-Laušević, S.N.; Božić, B.D.; Božić Nedeljković, B.D. Extracts Characterization and In Vitro Evaluation of Potential Immunomodulatory Activities of the Moss Hypnum cupressiforme Hedw. Molecules 2020, 25, 3343. [Google Scholar] [CrossRef]
- Lin, D.; Xiao, M.; Zhao, J.; Li, Z.; Xing, B.; Li, X.; Kong, M.; Li, L.; Zhang, Q.; Liu, Y.; et al. An Overview of Plant Phenolic Compounds and Their Importance in Human Nutrition and Management of Type 2 Diabetes. Molecules 2016, 21, 1374. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2016, gkw1000. [Google Scholar]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. In Structural Genomics; Springer: New York, NY, USA, 2021; pp. 239–255. [Google Scholar]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinforma. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Simoben, C.V.; Qaseem, A.; Moumbock, A.F.; Telukunta, K.K.; Günther, S.; Sippl, W.; Ntie-Kang, F. Pharmacoinformatic investigation of medicinal plants from East Africa. Mol. Inf. 2020, 39, 2000163. [Google Scholar] [CrossRef]
- Mumtaz, A.; Ashfaq, U.A.; ul Qamar, M.T.; Anwar, F.; Gulzar, F.; Ali, M.A.; Saari, N.; Pervez, M.T. MPD3: A useful medicinal plants database for drug designing. Nat. Prod. Res. 2017, 31, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E., III; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, P.; Vemulapalli, S.P.B.; Nath, N.; Griesinger, C.; Grubmüller, H. Probing the Accuracy of Explicit Solvent Constant pH Molecular Dynamics Simulations for Peptides. J. Chem. Theory Comput. 2020, 16, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Swails, J.; McGee, D., Jr. The Amber Project. Available online: https://ambermd.org/tutorials/advanced/tutorial18/section2.php (accessed on 21 December 2022).
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Mishra, S.K.; Koča, J. Assessing the Performance of MM/PBSA, MM/GBSA, and QM–MM/GBSA Approaches on Protein/Carbohydrate Complexes: Effect of Implicit Solvent Models, QM Methods, and Entropic Contributions. J. Phys. Chem. B 2018, 122, 8113–8121. [Google Scholar] [CrossRef]
- Vangone, A.; Schaarschmidt, J.; Koukos, P.; Geng, C.; Citro, N.; Trellet, M.E.; Xue, L.C.; Bonvin, A.M. Large-scale prediction of binding affinity in protein–small ligand complexes: The PRODIGY-LIG web server. Bioinformatics 2019, 35, 1585–1587. [Google Scholar] [CrossRef]
- Kiss, R.; Sandor, M.; Szalai, F.A. http://Mcule.com: A public web service for drug discovery. J Cheminform. 2012, 4, P17. [Google Scholar] [CrossRef]
- Yayıntaş, O.T.; Yılmaz, S.; Sökmen, M. Determination of antioxidant, antimicrobial and antitumor activity of bryophytes from Mount Ida (Canakkale, Turkey). Indian J. Tradit. Knowl. 2019, 18, 395–401. [Google Scholar]
- Sunghwa, F.; Koketsu, M. Phenolic and bis-iridoid glycosides from Strychnos cocculoides. Nat. Prod. Res. 2009, 23, 1408–1415. [Google Scholar] [CrossRef]
- Sitrit, Y.; Loison, S.; Ninio, R.; Dishon, E.; Bar, E.; Lewinsohn, E.; Mizrahi, Y. Characterization of monkey orange (Strychnos spinosa Lam.), a potential new crop for arid regions. J. Agric. Food Chem. 2003, 51, 6256–6260. [Google Scholar] [CrossRef]
- Mwamba, C.K. Monkey Orange: Strychnos cocculoides, 1st ed.; International Centre for Underutilised Crops, Southampton University: Southampton, UK, 2006. [Google Scholar]
- Genheden, S.; Ryde, U. How to obtain statistically converged MM/GBSA results. J. Comput. Chem. 2010, 31, 837–846. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2D Structure | Compound Name | IFD Score | Identifier |
|---|---|---|---|
 | 3,5,7,4′-Tetrahydroxy flavanone 3′-(4-hydroxybenzoic acid) | −11.54 | A |
 | 2-Hydroxy-3-O-beta-glucopyranosyl-benzoic acid | −10.0 | B |
 | 5,3′-Dihydroxy-7,4′-dimethoxyflavanone 3′-glucoside | −9.76 | C |
 | 5,7,3′-Trihydroxy-2′,4′-dimethoxy-6-prenylisoflavanone | −9.66 | D |
| Compound | Identifier | MW. (g/mol) | Source | Molecule Class | Biological Activity | Lipinski Violation | AMES * Toxicity | Rat Oral LD50 (mol/kg) | Max. Tolerated Dose (Human) (log Mg/kg/day) | T. pyriformis Toxicity ** (log µg/L) | HBD | HBA | Rotatable Bonds No. | TPSA | Bioactivity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3,5,7,4′-Tetrahydroxy flavanone 3′-(4-hydroxybenzoic acid) | A | 424.36 | Hypnum cupressiforme | Flavonoid | Anti-viral | 1 | - | - | - | - | 6 | 9 | 3 | 164.74 Å2 | 0.24 |
| 2-hydroxy-3-O-beta-glucopyranosyl-benzoic acid | B | 316.26 | Strychnos cocculoides | Phenolic | Anti-inflammatory | 1 | No | 2.305 | 1.294 | 0.285 | 6 | 9 | 4 | 156.91 Å2 | 0.43 |
| MM/GBSA | Compound A-Run 1 | Compound A-Run 2 | Compound A-Run 3 | Average | SD | Compound B-Run 1 | Compound B-Run 2 | Compound B-Run 3 | Average | SD |
|---|---|---|---|---|---|---|---|---|---|---|
| vdW | −71.16 | −68.44 | −64.37 | −68.0 | 3.4 | −50.96 | −50.96 | −51.67 | −51.2 | 0.41 |
| Electrostatic | −22.77 | −21.72 | −21.89 | −22.1 | 0.6 | −34.13 | −28.76 | −21.22 | −28.0 | 6.49 |
| ESURF | 23.29 | 18.78 | 17.64 | 19.9 | 3.0 | 28.10 | 24.52 | 23.78 | 25.5 | 2.31 |
| EGB | 8.05 | 10.01 | 8.85 | 9.0 | 1.0 | 13.83 | 9.29 | 4.78 | 9.3 | 4.53 |
| ΔG Bind | −62.59 | −61.37 | −59.77 | −61.2 | 1.4 | −43.16 | −47.09 | −44.33 | −44.9 | 2.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammad, A.; Alshawaf, E.; Arefanian, H.; Marafie, S.K.; Khan, A.; Wei, D.-Q.; Al-Mulla, F.; Abubaker, J. Targeting SARS-CoV-2 Macrodomain-1 to Restore the Innate Immune Response Using In Silico Screening of Medicinal Compounds and Free Energy Calculation Approaches. Viruses 2023, 15, 1907. https://doi.org/10.3390/v15091907
Mohammad A, Alshawaf E, Arefanian H, Marafie SK, Khan A, Wei D-Q, Al-Mulla F, Abubaker J. Targeting SARS-CoV-2 Macrodomain-1 to Restore the Innate Immune Response Using In Silico Screening of Medicinal Compounds and Free Energy Calculation Approaches. Viruses. 2023; 15(9):1907. https://doi.org/10.3390/v15091907
Chicago/Turabian StyleMohammad, Anwar, Eman Alshawaf, Hossein Arefanian, Sulaiman K. Marafie, Abbas Khan, Dong-Qing Wei, Fahd Al-Mulla, and Jehad Abubaker. 2023. "Targeting SARS-CoV-2 Macrodomain-1 to Restore the Innate Immune Response Using In Silico Screening of Medicinal Compounds and Free Energy Calculation Approaches" Viruses 15, no. 9: 1907. https://doi.org/10.3390/v15091907