
Current ARTs, Virologic Failure, and Implications for AIDS Management: A Systematic Review

Abstract
:1. Introduction
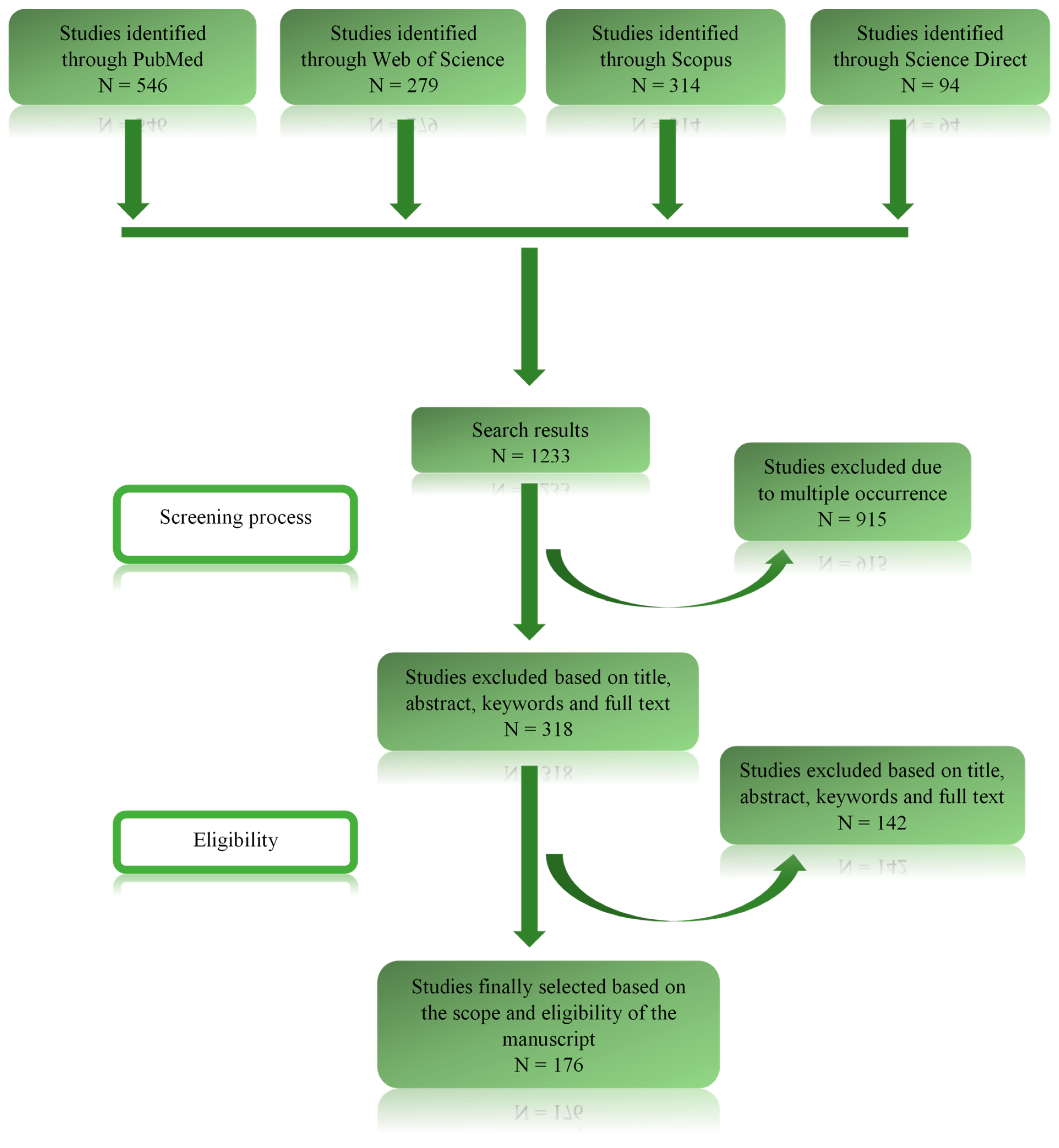
2. Methodology
2.1. Research Enquiry and Question Formulation
2.2. Databases Used for Data Retrieval and Evaluation
2.3. Criteria Determining the Study Selection
2.4. Data Analysis and Possible Sources of Bias
3. Results
4. Preliminary Analysis of the Data Retrieved from the Literature
4.1. Nucleoside/Nucleotide Reverse Transcriptase Inhibitors (NRTIs)
4.1.1. NRTIs Mode of Action
4.1.2. Metabolism and Side Effects of NRTIs
4.1.3. Mechanism of Resistance to NRTIs
4.2. Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs)
4.2.1. NNRTIs Mode of Action
4.2.2. Metabolism and Side Effects of NNRTIs
4.2.3. Mechanism of Resistance to NNRTIs
4.3. Protease Inhibitors (PIs)
4.3.1. PIs Mechanism of Action
4.3.2. Metabolism and Side Effects of PIs
4.3.3. Mechanism of Resistance to PIs
4.4. Integrase Strand Transfer Inhibitors (INSTIs)
4.4.1. INSTIs Mechanism of Action
4.4.2. Metabolism and Side Effects of INSTIs
4.4.3. Mechanism of Resistance to INSTIs
4.5. Entry Inhibitors (CCR5 Receptor Antagonists and Fusion Inhibitors)
4.5.1. Mode of Action of CCR5 Receptor Antagonists
4.5.2. Metabolism and Side Effects of Maraviroc
4.5.3. Mechanism of Resistance to CCR5 Receptor Antagonists
4.6. Other HIV-1 Virus Entry Inhibitors
4.6.1. Monoclonal-Antibody Based (mAb) Antiretroviral Therapy
4.6.2. Peptide-Based Inhibitors (Fusion Inhibitors)
4.6.3. Mechanism of Resistance to Ibalizumab
5. Drivers of Virologic Failure
5.1. Addressing Virologic Failure Resulting from Resistant Strains
5.2. Addressing Virologic Failure Resulting from Poor Adherence to ART Regimens
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Spending on Health and HIV/AIDS: Domestic Health Spending and Development Assistance in 188 Countries, 1995–2015. Lancet Lond. Engl. 2018, 391, 1799–1829. [CrossRef] [Green Version]
- The Joint United Nations Programme on HIV/AIDS (UNAIDS). Available online: https://www.unaids.org/en/resources/documents/2023/2022_unaids_data (accessed on 4 May 2023).
- Duncombe, C.; Ravishankar, S.; Zuniga, J.M. Fast-Track Cities: Striving to End Urban HIV Epidemics by 2030. Curr. Opin. HIV AIDS 2019, 14, 503–508. [Google Scholar] [CrossRef]
- Global, Regional, and National Incidence, Prevalence, and Mortality of HIV, 1980–2017, and Forecasts to 2030, for 195 Countries and Territories: A Systematic Analysis for the Global Burden of Diseases, Injuries, and Risk Factors Study 2017. Lancet HIV 2019, 6, e831–e859. [CrossRef] [Green Version]
- Kemnic, T.R.; Gulick, P.G. HIV Antiretroviral Therapy; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK513308/ (accessed on 6 April 2022).
- Cowan, E.A.; McGowan, J.P.; Fine, S.M.; Vail, R.M.; Merrick, S.T.; Radix, A.E.; Hoffmann, C.J.; Gonzalez, C.J. Diagnosis and Management of Acute HIV; Johns Hopkins University: Baltimore, MD, USA, 2021. [Google Scholar]
- World Health Organization (WHO). Consolidated Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing HIV Infection: Recommendations for a Public Health Approach, 2nd ed.; WHO: Geneva, Switzerland, 2016; Available online: https://apps.who.int/iris/handle/10665/208825 (accessed on 4 May 2023).
- NIH’s Office of AIDS Research. Available online: https://clinicalinfo.hiv.gov/en/glossary/virologic-failure (accessed on 5 May 2023).
- McCluskey, S.M.; Siedner, M.J.; Marconi, V.C. Management of Virologic Failure and HIV Drug Resistance. Infect. Dis. Clin. N. Am. 2019, 33, 707–742. [Google Scholar] [CrossRef]
- Mbhele, N.; Chimukangara, B.; Gordon, M. HIV-1 Integrase Strand Transfer Inhibitors: A Review of Current Drugs, Recent Advances and Drug Resistance. Int. J. Antimicrob. Agents 2021, 57, 106343. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA Statement for Reporting Systematic Reviews and Meta-Analyses of Studies That Evaluate Healthcare Interventions: Explanation and Elaboration. BMJ 2009, 339, b2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foka, F.E.T.; Manamela, N.; Mufamadi, S.M.; Mufhandu, H.T. Potential of Azadirachta Indica as a Capping Agent for Antiviral Nanoparticles against SARS-CoV-2. BioMed Res. Int. 2022, 2022, 5714035. [Google Scholar] [CrossRef] [PubMed]
- Kolata, G. FDA Approves AZT. Science 1987, 235, 1570. [Google Scholar] [CrossRef]
- Fauci, A.S. HIV and AIDS: 20 Years of Science. Nat. Med. 2003, 9, 839–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menéndez-Arias, L.; Delgado, R. Update and Latest Advances in Antiretroviral Therapy. Trends Pharmacol. Sci. 2022, 43, 16–29. [Google Scholar] [CrossRef]
- De Clercq, E. Anti-HIV Drugs: 25 Compounds Approved within 25 Years after the Discovery of HIV. Int. J. Antimicrob. Agents 2009, 33, 307–320. [Google Scholar] [CrossRef]
- Lee, F.J.; Amin, J.; Carr, A. Efficacy of Initial Antiretroviral Therapy for HIV-1 Infection in Adults: A Systematic Review and Meta-Analysis of 114 Studies with up to 144 Weeks’ Follow-Up. PLoS ONE 2014, 9, e97482. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.H.; Zulfiqar, H. Reverse Transcriptase Inhibitors; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK551504 (accessed on 11 March 2023).
- Richman, D.D.; Havlir, D.; Corbeil, J.; Looney, D.; Ignacio, C.; Spector, S.A.; Sullivan, J.; Cheeseman, S.; Barringer, K.; Pauletti, D. Nevirapine Resistance Mutations of Human Immunodeficiency Virus Type 1 Selected during Therapy. J. Virol. 1994, 68, 1660–1666. [Google Scholar] [CrossRef]
- Saravanan, S.; Kausalya, B.; Gomathi, S.; Sivamalar, S.; Pachamuthu, B.; Selvamuthu, P.; Pradeep, A.; Sunil, S.; Mothi, S.N.; Smith, D.M.; et al. Etravirine and Rilpivirine Drug Resistance Among HIV-1 Subtype C Infected Children Failing Non-Nucleoside Reverse Transcriptase Inhibitor-Based Regimens in South India. AIDS Res. Hum. Retrovir. 2017, 33, 567–574. [Google Scholar] [CrossRef]
- Diphoko, T.; Gaseitsiwe, S.; Kasvosve, I.; Moyo, S.; Okatch, H.; Musonda, R.; Wainberg, M.; Makhema, J.; Marlink, R.; Novitsky, V.; et al. Prevalence of Rilpivirine and Etravirine Resistance Mutations in HIV-1 Subtype C-Infected Patients Failing Nevirapine or Efavirenz-Based Combination Antiretroviral Therapy in Botswana. AIDS Res. Hum. Retrovir. 2018, 34, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Stanford University HIV Drug Resistance Database. Available online: https://hivdb.stanford.edu/dr-summary/resistance-notes/NNRTI/ (accessed on 11 March 2023).
- Cahn, P.; Sax, P.E.; Squires, K.; Molina, J.-M.; Ratanasuwan, W.; Rassool, M.; Bloch, M.; Xu, X.; Zhou, Y.; Homony, B.; et al. Raltegravir 1200 Mg Once Daily vs 400 Mg Twice Daily, With Emtricitabine and Tenofovir Disoproxil Fumarate, for Previously Untreated HIV-1 Infection: Week 96 Results From ONCEMRK, a Randomized, Double-Blind, Noninferiority Trial. J. Acquir. Immune Defic. Syndr. 2018, 78, 589–598. [Google Scholar] [CrossRef]
- Quashie, P.K.; Mesplède, T.; Wainberg, M.A. HIV Drug Resistance and the Advent of Integrase Inhibitors. Curr. Infect. Dis. Rep. 2013, 15, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Chu, Y.; Wang, Y. HIV Protease Inhibitors: A Review of Molecular Selectivity and Toxicity. HIV/AIDS Res. Palliat. Care 2015, 7, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Alencar, C.S.; Nishiya, A.S.; Ferreira, S.; Giret, M.T.M.; Diaz, R.S.; Sabino, E.C. Evaluation of Primary Resistance to HIV Entry Inhibitors among Brazilian Patients Failing Reverse Transcriptase/Protease Inhibitors Treatment Reveal High Prevalence of Maraviroc Resistance-Related Mutations. AIDS Res. Hum. Retrovir. 2010, 26, 1267–1271. [Google Scholar] [CrossRef]
- Zhao, A.V.; Crutchley, R.D.; Guduru, R.C.; Ton, K.; Lam, T.; Min, A.C. A Clinical Review of HIV Integrase Strand Transfer Inhibitors (INSTIs) for the Prevention and Treatment of HIV-1 Infection. Retrovirology 2022, 19, 22. [Google Scholar] [CrossRef]
- Arribas, J.R.; Thompson, M.; Sax, P.E.; Haas, B.; McDonald, C.; Wohl, D.A.; DeJesus, E.; Clarke, A.E.; Guo, S.; Wang, H.; et al. Brief Report: Randomized, Double-Blind Comparison of Tenofovir Alafenamide (TAF) vs Tenofovir Disoproxil Fumarate (TDF), Each Coformulated with Elvitegravir, Cobicistat, and Emtricitabine (E/C/F) for Initial HIV-1 Treatment: Week 144 Results. J. Acquir. Immune Defic. Syndr. 2017, 75, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Malet, I.; Delelis, O.; Valantin, M.-A.; Montes, B.; Soulie, C.; Wirden, M.; Tchertanov, L.; Peytavin, G.; Reynes, J.; Mouscadet, J.-F.; et al. Mutations Associated with Failure of Raltegravir Treatment Affect Integrase Sensitivity to the Inhibitor in Vitro. Antimicrob. Agents Chemother. 2008, 52, 1351–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malet, I.; Subra, F.; Charpentier, C.; Collin, G.; Descamps, D.; Calvez, V.; Marcelin, A.-G.; Delelis, O. Mutations Located Outside the Integrase Gene Can Confer Resistance to HIV-1 Integrase Strand Transfer Inhibitors. mBio 2017, 8, e00922-17. [Google Scholar] [CrossRef] [Green Version]
- Wilkin, T.J.; Gulick, R.M. CCR5 Antagonism in HIV Infection: Current Concepts and Future Opportunities. Annu. Rev. Med. 2012, 63, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Yost, R.; Pasquale, T.R.; Sahloff, E.G. Maraviroc: A Coreceptor CCR5 Antagonist for Management of HIV Infection. Am. J. Health Syst. Pharm. 2009, 66, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Woollard, S.M.; Kanmogne, G.D. Maraviroc: A Review of Its Use in HIV Infection and Beyond. Drug Des. Devel. Ther. 2015, 9, 5447–5468. [Google Scholar] [CrossRef] [Green Version]
- Fätkenheuer, G.; Nelson, M.; Lazzarin, A.; Konourina, I.; Hoepelman, A.I.M.; Lampiris, H.; Hirschel, B.; Tebas, P.; Raffi, F.; Trottier, B.; et al. Subgroup Analyses of Maraviroc in Previously Treated R5 HIV-1 Infection. N. Engl. J. Med. 2008, 359, 1442–1455. [Google Scholar] [CrossRef]
- Swenson, L.C.; Chui, C.K.S.; Brumme, C.J.; Chan, D.; Woods, C.K.; Mo, T.; Dong, W.; Chapman, D.; Lewis, M.; Demarest, J.F.; et al. Genotypic Analysis of the V3 Region of HIV from Virologic Nonresponders to Maraviroc-Containing Regimens Reveals Distinct Patterns of Failure. Antimicrob. Agents Chemother. 2013, 57, 6122–6130. [Google Scholar] [CrossRef] [Green Version]
- Xiao, T.; Cai, Y.; Chen, B. HIV-1 Entry and Membrane Fusion Inhibitors. Viruses 2021, 13, 735. [Google Scholar] [CrossRef]
- Awi, N.J.; Teow, S.-Y. Antibody-Mediated Therapy against HIV/AIDS: Where Are We Standing Now? J. Pathog. 2018, 2018, 8724549. [Google Scholar] [CrossRef] [PubMed]
- Miner, M.D.; Corey, L.; Montefiori, D. Broadly Neutralizing Monoclonal Antibodies for HIV Prevention. J. Int. AIDS Soc. 2021, 24 (Suppl. S7), e25829. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Dimitrov, D.S. Monoclonal Antibody-Based Candidate Therapeutics against HIV Type 1. AIDS Res. Hum. Retrovir. 2012, 28, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holec, A.D.; Mandal, S.; Prathipati, P.K.; Destache, C.J. Nucleotide Reverse Transcriptase Inhibitors: A Thorough Review, Present Status and Future Perspective as HIV Therapeutics. Curr. HIV Res. 2017, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.S.; Olson, L.; Fridland, A. Role of Purine Nucleoside Phosphorylase in Interactions between 2′,3′-Dideoxyinosine and Allopurinol, Ganciclovir, or Tenofovir. Antimicrob. Agents Chemother. 2004, 48, 1089–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masereeuw, R.; Russel, F.G.M. Regulatory Pathways for ATP-Binding Cassette Transport Proteins in Kidney Proximal Tubules. AAPS J. 2012, 14, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Furukawa, T.; Nitanda, T.; Okamoto, M.; Sugimoto, Y.; Akiyama, S.-I.; Baba, M. Breast Cancer Resistance Protein (BCRP/ABCG2) Induces Cellular Resistance to HIV-1 Nucleoside Reverse Transcriptase Inhibitors. Mol. Pharmacol. 2003, 63, 65–72. [Google Scholar] [CrossRef]
- Woodward, C.L.N.; Hall, A.M.; Williams, I.G.; Madge, S.; Copas, A.; Nair, D.; Edwards, S.G.; Johnson, M.A.; Connolly, J.O. Tenofovir-Associated Renal and Bone Toxicity. HIV Med. 2009, 10, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Kohler, J.J.; Lewis, W. A Brief Overview of Mechanisms of Mitochondrial Toxicity from NRTIs. Environ. Mol. Mutagen. 2007, 48, 166–172. [Google Scholar] [CrossRef]
- Rosenfeldt, F.L.; Mijch, A.; McCrystal, G.; Sweeney, C.; Pepe, S.; Nicholls, M.; Dennett, X. Skeletal Myopathy Associated with Nucleoside Reverse Transcriptase Inhibitor Therapy: Potential Benefit of Coenzyme Q10 Therapy. Int. J. STD AIDS 2005, 16, 827–829. [Google Scholar] [CrossRef]
- Calza, L.; Manfredi, R.; Chiodo, F. Hyperlactataemia and Lactic Acidosis in HIV-Infected Patients Receiving Antiretroviral Therapy. Clin. Nutr. Edinb. Scotl. 2005, 24, 5–15. [Google Scholar] [CrossRef]
- Segarra-Newnham, M.; Soffler, S.L. Osteoporosis and Vitamin D Deficiency in HIV-Positive Patients. J. Pharm. Technol. 2011, 27, 251–257. [Google Scholar] [CrossRef]
- Custodio, J.M.; Fordyce, M.; Garner, W.; Vimal, M.; Ling, K.H.J.; Kearney, B.P.; Ramanathan, S. Pharmacokinetics and Safety of Tenofovir Alafenamide in HIV-Uninfected Subjects with Severe Renal Impairment. Antimicrob. Agents Chemother. 2016, 60, 5135–5140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, N.; Shaik, N.; Pan, G.; Terasaki, T.; Mukai, C.; Kitagaki, S.; Miyakoshi, N.; Elmquist, W.F. Investigation of the Role of Breast Cancer Resistance Protein (Bcrp/Abcg2) on Pharmacokinetics and Central Nervous System Penetration of Abacavir and Zidovudine in the Mouse. Drug Metab. Dispos. Biol. Fate Chem. 2008, 36, 1476–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arts, E.J.; Hazuda, D.J. HIV-1 Antiretroviral Drug Therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [Green Version]
- Regina, G.L.; Coluccia, A.; Silvestri, R. Looking for an Active Conformation of the Future HIV Type-1 Non-Nucleoside Reverse Transcriptase Inhibitors. Antivir. Chem. Chemother. 2010, 20, 213–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sluis-Cremer, N.; Tachedjian, G. Mechanisms of Inhibition of HIV Replication by Non-Nucleoside Reverse Transcriptase Inhibitors. Virus Res. 2008, 134, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Usach, I.; Melis, V.; Peris, J.-E. Non-Nucleoside Reverse Transcriptase Inhibitors: A Review on Pharmacokinetics, Pharmacodynamics, Safety and Tolerability. J. Int. AIDS Soc. 2013, 16, 18567. [Google Scholar] [CrossRef]
- Marzinke, M.A. Chapter 6—Therapeutic Drug Monitoring of Antiretrovirals. In Clinical Challenges in Therapeutic Drug Monitoring; Clarke, W., Dasgupta, A., Eds.; Elsevier: San Diego, CA, USA, 2016; pp. 135–163. ISBN 978-0-12-802025-8. [Google Scholar]
- Erickson, D.A.; Mather, G.; Trager, W.F.; Levy, R.H.; Keirns, J.J. Characterization of the in Vitro Biotransformation of the HIV-1 Reverse Transcriptase Inhibitor Nevirapine by Human Hepatic Cytochromes P-450. Drug Metab. Dispos. Biol. Fate Chem. 1999, 27, 1488–1495. [Google Scholar]
- von Moltke, L.L.; Greenblatt, D.J.; Granda, B.W.; Giancarlo, G.M.; Duan, S.X.; Daily, J.P.; Harmatz, J.S.; Shader, R.I. Inhibition of Human Cytochrome P450 Isoforms by Nonnucleoside Reverse Transcriptase Inhibitors. J. Clin. Pharmacol. 2001, 41, 85–91. [Google Scholar] [CrossRef]
- Podzamczer, D.; Fumero, E. The Role of Nevirapine in the Treatment of HIV-1 Disease. Expert Opin. Pharmacother. 2001, 2, 2065–2078. [Google Scholar] [CrossRef]
- Best, B.M.; Goicoechea, M. Efavirenz--Still First-Line King? Expert Opin. Drug Metab. Toxicol. 2008, 4, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, M.S.; Günthard, H.F.; Schapiro, J.M.; Brun-Vézinet, F.; Clotet, B.; Hammer, S.M.; Johnson, V.A.; Kuritzkes, D.R.; Mellors, J.W.; Pillay, D.; et al. Antiretroviral Drug Resistance Testing in Adult HIV-1 Infection: 2008 Recommendations of an International AIDS Society-USA Panel. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2008, 47, 266–285. [Google Scholar] [CrossRef] [Green Version]
- Nastri, B.M.; Pagliano, P.; Zannella, C.; Folliero, V.; Masullo, A.; Rinaldi, L.; Galdiero, M.; Franci, G. HIV and Drug-Resistant Subtypes. Microorganisms 2023, 11, 221. [Google Scholar] [CrossRef]
- Malaty, L.I.; Kuper, J.J. Drug Interactions of HIV Protease Inhibitors. Drug Saf. 1999, 20, 147–169. [Google Scholar] [CrossRef]
- Hruz, P.W. HIV Protease Inhibitors and Insulin Resistance: Lessons from in-Vitro, Rodent and Healthy Human Volunteer Models. Curr. Opin. HIV AIDS 2008, 3, 660–665. [Google Scholar] [CrossRef] [Green Version]
- Bozzette, S.A.; Ake, C.F.; Tam, H.K.; Chang, S.W.; Louis, T.A. Cardiovascular and Cerebrovascular Events in Patients Treated for Human Immunodeficiency Virus Infection. N. Engl. J. Med. 2003, 348, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Soontornniyomkij, V.; Umlauf, A.; Chung, S.A.; Cochran, M.L.; Soontornniyomkij, B.; Gouaux, B.; Toperoff, W.; Moore, D.J.; Masliah, E.; Ellis, R.J.; et al. HIV Protease Inhibitor Exposure Predicts Cerebral Small Vessel Disease. AIDS 2014, 28, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Karunaianantham, R.; Nesa Kumar, M.; Gopalan, B.; Haribabu, H.; Hanna, L.E.; Sanjeeva, G.N.; Reddy, D.; Shet, A.; Swaminathan, S.; Padmapriyadarsini, C. Molecular Characterization of the Pol Gene of Vertically Transmitted HIV-1 Strains in Children with Virological Failure. AIDS Res. Hum. Retrovir. 2022, 38, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.M.; Perno, C.F. HIV-1 Genetic Variability and Clinical Implications. Int. Sch. Res. Not. 2013, 2013, 481314. [Google Scholar] [CrossRef] [Green Version]
- Obasa, A.E.; Mikasi, S.G.; Brado, D.; Cloete, R.; Singh, K.; Neogi, U.; Jacobs, G.B. Drug Resistance Mutations Against Protease, Reverse Transcriptase and Integrase Inhibitors in People Living With HIV-1 Receiving Boosted Protease Inhibitors in South Africa. Front. Microbiol. 2020, 11, 438. [Google Scholar] [CrossRef] [Green Version]
- Podany, A.T.; Scarsi, K.K.; Pham, M.M.; Fletcher, C.V. Comparative Clinical Pharmacokinetics and Pharmacodynamics of HIV-1 Integrase Strand Transfer Inhibitors: An Updated Review. Clin. Pharmacokinet. 2020, 59, 1085–1107. [Google Scholar] [CrossRef]
- Reese, M.J.; Savina, P.M.; Generaux, G.T.; Tracey, H.; Humphreys, J.E.; Kanaoka, E.; Webster, L.O.; Harmon, K.A.; Clarke, J.D.; Polli, J.W. In Vitro Investigations into the Roles of Drug Transporters and Metabolizing Enzymes in the Disposition and Drug Interactions of Dolutegravir, a HIV Integrase Inhibitor. Drug Metab. Dispos. Biol. Fate Chem. 2013, 41, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Wensing, A.M.J.; Fun, A.; Nijhuis, M. HIV Protease Inhibitor Resistance. In Handbook of Antimicrobial Resistance; Berghuis, A., Matlashewski, G., Wainberg, M.A., Sheppard, D., Gotte, M., Eds.; Springer: New York, NY, USA, 2017; pp. 567–602. ISBN 978-1-4939-0694-9. [Google Scholar]
- Huang, W.; Frantzell, A.; Fransen, S.; Petropoulos, C.J. Multiple Genetic Pathways Involving Amino Acid Position 143 of HIV-1 Integrase Are Preferentially Associated with Specific Secondary Amino Acid Substitutions and Confer Resistance to Raltegravir and Cross-Resistance to Elvitegravir. Antimicrob. Agents Chemother. 2013, 57, 4105–4113. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.Z.; Smith, S.J.; Métifiot, M.; Marchand, C.; Boyer, P.L.; Pommier, Y.; Hughes, S.H.; Burke, T.R.J. 4-Amino-1-Hydroxy-2-Oxo-1,8-Naphthyridine-Containing Compounds Having High Potency against Raltegravir-Resistant Integrase Mutants of HIV-1. J. Med. Chem. 2014, 57, 5190–5202. [Google Scholar] [CrossRef] [PubMed]
- Wohl, D.A.; Cohen, C.; Gallant, J.E.; Mills, A.; Sax, P.E.; Dejesus, E.; Zolopa, A.; Liu, H.C.; Plummer, A.; White, K.L.; et al. A Randomized, Double-Blind Comparison of Single-Tablet Regimen Elvitegravir/Cobicistat/Emtricitabine/Tenofovir DF versus Single-Tablet Regimen Efavirenz/Emtricitabine/Tenofovir DF for Initial Treatment of HIV-1 Infection: Analysis of Week 144 Results. J. Acquir. Immune Defic. Syndr. 2014, 65, e118–e120. [Google Scholar] [CrossRef] [PubMed]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell Binding and Entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, G.; Combadiere, C.; Broder, C.C.; Feng, Y.; Kennedy, P.E.; Murphy, P.M.; Berger, E.A. CC CKR5: A RANTES, MIP-1alpha, MIP-1beta Receptor as a Fusion Cofactor for Macrophage-Tropic HIV-1. Science 1996, 272, 1955–1958. [Google Scholar] [CrossRef]
- Nichols, W.G.; Steel, H.M.; Bonny, T.; Adkison, K.; Curtis, L.; Millard, J.; Kabeya, K.; Clumeck, N. Hepatotoxicity Observed in Clinical Trials of Aplaviroc (GW873140). Antimicrob. Agents Chemother. 2008, 52, 858–865. [Google Scholar] [CrossRef] [Green Version]
- Wilkin, T.J.; Su, Z.; Krambrink, A.; Long, J.; Greaves, W.; Gross, R.; Hughes, M.D.; Flexner, C.; Skolnik, P.R.; Coakley, E.; et al. Three-Year Safety and Efficacy of Vicriviroc, a CCR5 Antagonist, in HIV-1-Infected Treatment-Experienced Patients. J. Acquir. Immune Defic. Syndr. 2010, 54, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Anstee, Q.M.; Neuschwander-Tetri, B.A.; Wai-Sun Wong, V.; Abdelmalek, M.F.; Rodriguez-Araujo, G.; Landgren, H.; Park, G.S.; Bedossa, P.; Alkhouri, N.; Tacke, F.; et al. Cenicriviroc Lacked Efficacy to Treat Liver Fibrosis in Nonalcoholic Steatohepatitis: AURORA Phase III Randomized Study. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Abel, S.; Russell, D.; Whitlock, L.A.; Ridgway, C.E.; Nedderman, A.N.R.; Walker, D.K. Assessment of the Absorption, Metabolism and Absolute Bioavailability of Maraviroc in Healthy Male Subjects. Br. J. Clin. Pharmacol. 2008, 65 (Suppl. S1), 60–67. [Google Scholar] [CrossRef] [Green Version]
- Araújo, L.A.L.; Almeida, S.E.M. HIV-1 Diversity in the Envelope Glycoproteins: Implications for Viral Entry Inhibition. Viruses 2013, 5, 595–604. [Google Scholar] [CrossRef] [Green Version]
- Araújo, L.A.L.; Junqueira, D.M.; de Medeiros, R.M.; Matte, M.C.C.; de Matos Almeida, S.E. Naturally Occurring Resistance Mutations to HIV-1 Entry Inhibitors in Subtypes B, C, and CRF31_BC. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2012, 54, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Eggink, D.; Bontjer, I.; de Taeye, S.W.; Langedijk, J.P.M.; Berkhout, B.; Sanders, R.W. HIV-1 Anchor Inhibitors and Membrane Fusion Inhibitors Target Distinct but Overlapping Steps in Virus Entry. J. Biol. Chem. 2019, 294, 5736–5746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, S.; Gondwe, C.; Tully, D.C.; Minhas, V.; Shea, D.; Kankasa, C.; M’soka, T.; Wood, C. Short Communication: Antiretroviral Therapy Resistance Mutations Present in the HIV Type 1 Subtype C Pol and Env Regions from Therapy-Naive Patients in Zambia. AIDS Res. Hum. Retrovir. 2010, 26, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Freeman, M.M.; Seaman, M.S.; Rits-Volloch, S.; Hong, X.; Kao, C.-Y.; Ho, D.D.; Chen, B. Crystal Structure of HIV-1 Primary Receptor CD4 in Complex with a Potent Antiviral Antibody. Structure 2010, 18, 1632–1641. [Google Scholar] [CrossRef] [Green Version]
- Acharya, P.; Luongo, T.S.; Louder, M.K.; McKee, K.; Yang, Y.; Kwon, Y.D.; Mascola, J.R.; Kessler, P.; Martin, L.; Kwong, P.D. Structural Basis for Highly Effective HIV-1 Neutralization by CD4-Mimetic Miniproteins Revealed by 1.5 Å Cocrystal Structure of Gp120 and M48U1. Structure 2013, 21, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Lu, L.; Liu, Q.; Yu, X.; Wang, L.; He, E.; Zou, P.; Du, L.; Sanders, R.W.; Liu, S.; et al. ADS-J1 Inhibits HIV-1 Infection and Membrane Fusion by Targeting the Highly Conserved Pocket in the Gp41 NHR-Trimer. Biochim. Biophys. Acta 2014, 1838, 1296–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, G.; Rits-Volloch, S.; Zhang, X.-Q.; Schooley, R.T.; Chen, B.; Harrison, S.C. Small Molecules That Bind the Inner Core of Gp41 and Inhibit HIV Envelope-Mediated Fusion. Proc. Natl. Acad. Sci. USA 2006, 103, 13938–13943. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lu, H.; Hou, L.; Qi, Z.; Teixeira, C.; Barbault, F.; Fan, B.-T.; Liu, S.; Jiang, S.; Xie, L. Design, Synthesis, and Biological Evaluation of N-Carboxyphenylpyrrole Derivatives as Potent HIV Fusion Inhibitors Targeting Gp41. J. Med. Chem. 2008, 51, 7843–7854. [Google Scholar] [CrossRef] [Green Version]
- Alam, S.M.; Morelli, M.; Dennison, S.M.; Liao, H.-X.; Zhang, R.; Xia, S.-M.; Rits-Volloch, S.; Sun, L.; Harrison, S.C.; Haynes, B.F.; et al. Role of HIV Membrane in Neutralization by Two Broadly Neutralizing Antibodies. Proc. Natl. Acad. Sci. USA 2009, 106, 20234–20239. [Google Scholar] [CrossRef]
- Chen, J.; Frey, G.; Peng, H.; Rits-Volloch, S.; Garrity, J.; Seaman, M.S.; Chen, B. Mechanism of HIV-1 Neutralization by Antibodies Targeting a Membrane-Proximal Region of Gp41. J. Virol. 2014, 88, 1249–1258. [Google Scholar] [CrossRef] [Green Version]
- Pelegrin, M.; Naranjo-Gomez, M.; Piechaczyk, M. Antiviral Monoclonal Antibodies: Can They Be More Than Simple Neutralizing Agents? Trends Microbiol. 2015, 23, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Ozorowski, G.; Pallesen, J.; de Val, N.; Lyumkis, D.; Cottrell, C.A.; Torres, J.L.; Copps, J.; Stanfield, R.L.; Cupo, A.; Pugach, P.; et al. Open and Closed Structures Reveal Allostery and Pliability in the HIV-1 Envelope Spike. Nature 2017, 547, 360–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pancera, M.; Lai, Y.-T.; Bylund, T.; Druz, A.; Narpala, S.; O’Dell, S.; Schön, A.; Bailer, R.T.; Chuang, G.-Y.; Geng, H.; et al. Crystal Structures of Trimeric HIV Envelope with Entry Inhibitors BMS-378806 and BMS-626529. Nat. Chem. Biol. 2017, 13, 1115–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilby, J.M.; Eron, J.J. Novel Therapies Based on Mechanisms of HIV-1 Cell Entry. N. Engl. J. Med. 2003, 348, 2228–2238. [Google Scholar] [CrossRef] [Green Version]
- Fessel, W.J.; Anderson, B.; Follansbee, S.E.; Winters, M.A.; Lewis, S.T.; Weinheimer, S.P.; Petropoulos, C.J.; Shafer, R.W. The Efficacy of an Anti-CD4 Monoclonal Antibody for HIV-1 Treatment. Antivir. Res. 2011, 92, 484–487. [Google Scholar] [CrossRef] [Green Version]
- Morellato-Castillo, L.; Acharya, P.; Combes, O.; Michiels, J.; Descours, A.; Ramos, O.H.P.; Yang, Y.; Vanham, G.; Ariën, K.K.; Kwong, P.D.; et al. Interfacial Cavity Filling to Optimize CD4–Mimetic Miniprotein Interactions with HIV-1 Surface Glycoprotein. J. Med. Chem. 2013, 56, 5033–5047. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Ofek, G.; Laub, L.; Louder, M.K.; Doria-Rose, N.A.; Longo, N.S.; Imamichi, H.; Bailer, R.T.; Chakrabarti, B.; Sharma, S.K.; et al. Broad and Potent Neutralization of HIV-1 by a Gp41-Specific Human Antibody. Nature 2012, 491, 406–412. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Cai, L.; Lu, H.; Qi, Z.; Jiang, S. Combinations of the First and Next Generations of Human Immunodeficiency Virus (HIV) Fusion Inhibitors Exhibit a Highly Potent Synergistic Effect against Enfuvirtide- Sensitive and -Resistant HIV Type 1 Strains. J. Virol. 2009, 83, 7862–7872. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Pan, C.; Xu, L.; Shui, Y.; Liu, K.; Jiang, S. Interactions between Different Generation HIV-1 Fusion Inhibitors and the Putative Mechanism Underlying the Synergistic Anti-HIV-1 Effect Resulting from Their Combination. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 1018–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Xiao, Y.; Song, H.; Liang, Q.; Ju, D.; Chen, X.; Lu, H.; Jing, W.; Jiang, S.; Zhang, L. Design and Evaluation of Sifuvirtide, a Novel HIV-1 Fusion Inhibitor. J. Biol. Chem. 2008, 283, 11126–11134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, B.; Yao, C.; Zhao, Q.-X.; Cai, W.-P.; Wang, M.; Lu, H.-Z.; Chen, Y.-Y.; Liu, L.; Wang, H.; He, Y.; et al. Efficacy and Safety of the Long-Acting Fusion Inhibitor Albuvirtide in Antiretroviral-Experienced Adults with Human Immunodeficiency Virus-1: Interim Analysis of the Randomized, Controlled, Phase 3, Non-Inferiority TALENT Study. Chin. Med. J. 2020, 133, 2919–2927. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Yao, X.; Zhang, C.; Cai, L.; Cui, S.; Wang, Y.; He, Y. Biophysical Property and Broad Anti-HIV Activity of Albuvirtide, a 3-Maleimimidopropionic Acid-Modified Peptide Fusion Inhibitor. PLoS ONE 2012, 7, e32599. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Ma, L.; Jiang, S.; Lu, H.; Liu, S.; He, Y.; Strick, N.; Neamati, N.; Debnath, A.K. Identification of N-Phenyl-N′-(2,2,6,6-Tetramethyl-Piperidin-4-Yl)-Oxalamides as a New Class of HIV-1 Entry Inhibitors That Prevent Gp120 Binding to CD4. Virology 2005, 339, 213–225. [Google Scholar] [CrossRef]
- Curreli, F.; Kwon, Y.D.; Zhang, H.; Scacalossi, D.; Belov, D.S.; Tikhonov, A.A.; Andreev, I.A.; Altieri, A.; Kurkin, A.V.; Kwong, P.D.; et al. Structure-Based Design of a Small Molecule CD4-Antagonist with Broad Spectrum Anti-HIV-1 Activity. J. Med. Chem. 2015, 58, 6909–6927. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhang, Z.; Wallace, O.B.; Deshpande, M.; Fang, H.; Yang, Z.; Zadjura, L.M.; Tweedie, D.L.; Huang, S.; Zhao, F.; et al. Discovery of 4-Benzoyl-1-[(4-Methoxy-1H- Pyrrolo[2,3-b]Pyridin-3-Yl)Oxoacetyl]-2- (R)-Methylpiperazine (BMS-378806): A Novel HIV-1 Attachment Inhibitor That Interferes with CD4-Gp120 Interactions. J. Med. Chem. 2003, 46, 4236–4239. [Google Scholar] [CrossRef]
- Hanna, G.J.; Lalezari, J.; Hellinger, J.A.; Wohl, D.A.; Nettles, R.; Persson, A.; Krystal, M.; Lin, P.; Colonno, R.; Grasela, D.M. Antiviral Activity, Pharmacokinetics, and Safety of BMS-488043, a Novel Oral Small-Molecule HIV-1 Attachment Inhibitor, in HIV-1-Infected Subjects. Antimicrob. Agents Chemother. 2011, 55, 722–728. [Google Scholar] [CrossRef] [Green Version]
- Kozal, M.; Aberg, J.; Pialoux, G.; Cahn, P.; Thompson, M.; Molina, J.-M.; Grinsztejn, B.; Diaz, R.; Castagna, A.; Kumar, P.; et al. Fostemsavir in Adults with Multidrug-Resistant HIV-1 Infection. N. Engl. J. Med. 2020, 382, 1232–1243. [Google Scholar] [CrossRef]
- Stiegler, G.; Kunert, R.; Purtscher, M.; Wolbank, S.; Voglauer, R.; Steindl, F.; Katinger, H. A Potent Cross-Clade Neutralizing Human Monoclonal Antibody against a Novel Epitope on Gp41 of Human Immunodeficiency Virus Type 1. AIDS Res. Hum. Retroviruses 2001, 17, 1757–1765. [Google Scholar] [CrossRef]
- Williams, L.D.; Ofek, G.; Schätzle, S.; McDaniel, J.R.; Lu, X.; Nicely, N.I.; Wu, L.; Lougheed, C.S.; Bradley, T.; Louder, M.K.; et al. Potent and Broad HIV-Neutralizing Antibodies in Memory B Cells and Plasma. Sci. Immunol. 2017, 2, eaal2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzo, S.; Castagna, A.; Monachetti, A.; Hasson, H.; Danise, A.; Carini, E.; Bagnarelli, P.; Lazzarin, A.; Clementi, M. Genotype and Phenotype Patterns of Human Immunodeficiency Virus Type 1 Resistance to Enfuvirtide during Long-Term Treatment. Antimicrob. Agents Chemother. 2004, 48, 3253–3259. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Chong, H.; Qiu, Z.; Xiong, S.; He, Y. Mechanism of HIV-1 Resistance to Short-Peptide Fusion Inhibitors Targeting the Gp41 Pocket. J. Virol. 2015, 89, 5801–5811. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Zhao, J.; Guo, F.; Ji, H.; Zhang, L.; Jiang, X.; Wang, L. HIV-1 Gp41 Genetic Diversity and Enfuvirtide Resistance-Associated Mutations among Enfuvirtide-Naïve Patients in Southern China. Virus Res. 2021, 292, 198215. [Google Scholar] [CrossRef] [PubMed]
- Beccari, M.V.; Mogle, B.T.; Sidman, E.F.; Mastro, K.A.; Asiago-Reddy, E.; Kufel, W.D. Ibalizumab, a Novel Monoclonal Antibody for the Management of Multidrug-Resistant HIV-1 Infection. Antimicrob. Agents Chemother. 2019, 63, e00110-19. [Google Scholar] [CrossRef] [Green Version]
- Khanlou, H.; Gathe, J.; Schrader, S.; Towner, W.; Weinheimer, S.; Lewis, S. Safety, Efficacy, and Pharmacokinetics of Ibalizumab in Treatment-Experienced HIV-1 Infected Patients: A Phase 2b Study. In Proceedings of the 51st Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 17–20 September 2011. [Google Scholar]
- Toma, J.; Weinheimer, S.P.; Stawiski, E.; Whitcomb, J.M.; Lewis, S.T.; Petropoulos, C.J.; Huang, W. Loss of Asparagine-Linked Glycosylation Sites in Variable Region 5 of Human Immunodeficiency Virus Type 1 Envelope Is Associated with Resistance to CD4 Antibody Ibalizumab. J. Virol. 2011, 85, 3872–3880. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-Q.; Sorensen, M.; Fung, M.; Schooley, R.T. Synergistic in Vitro Antiretroviral Activity of a Humanized Monoclonal Anti-CD4 Antibody (TNX-355) and Enfuvirtide (T-20). Antimicrob. Agents Chemother. 2006, 50, 2231–2233. [Google Scholar] [CrossRef] [Green Version]
- Gnanakaran, S.; Lang, D.; Daniels, M.; Bhattacharya, T.; Derdeyn, C.A.; Korber, B. Clade-Specific Differences between Human Immunodeficiency Virus Type 1 Clades B and C: Diversity and Correlations in C3-V4 Regions of Gp120. J. Virol. 2007, 81, 4886–4891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, M.C.; Goldie, S.J.; Losina, E.; Cohen, C.J.; Baxter, J.D.; Zhang, H.; Kimmel, A.D.; Freedberg, K.A. Use of Genotypic Resistance Testing to Guide HIV Therapy: Clinical Impact and Cost-Effectiveness. Ann. Intern. Med. 2001, 134, 440–450. [Google Scholar] [CrossRef]
- Dunn, D.T.; Green, H.; Loveday, C.; Rinehart, A.; Pillay, D.; Fisher, M.; McCormack, S.; Babiker, A.G.; Darbyshire, J.H. A Randomized Controlled Trial of the Value of Phenotypic Testing in Addition to Genotypic Testing for HIV Drug Resistance: Evaluation of Resistance Assays (ERA) Trial Investigators. J. Acquir. Immune Defic. Syndr. 2005, 38, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Durant, J.; Clevenbergh, P.; Halfon, P.; Delgiudice, P.; Porsin, S.; Simonet, P.; Montagne, N.; Boucher, C.A.; Schapiro, J.M.; Dellamonica, P. Drug-Resistance Genotyping in HIV-1 Therapy: The VIRADAPT Randomised Controlled Trial. Lancet 1999, 353, 2195–2199. [Google Scholar] [CrossRef]
- Baxter, J.D.; Mayers, D.L.; Wentworth, D.N.; Neaton, J.D.; Hoover, M.L.; Winters, M.A.; Mannheimer, S.B.; Thompson, M.A.; Abrams, D.I.; Brizz, B.J.; et al. A Randomized Study of Antiretroviral Management Based on Plasma Genotypic Antiretroviral Resistance Testing in Patients Failing Therapy. CPCRA 046 Study Team for the Terry Beirn Community Programs for Clinical Research on AIDS. AIDS 2000, 14, F83–F93. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, A.; Antinori, A.; Rizzo, M.G.; Murri, R.; Ammassari, A.; Baldini, F.; Di Giambenedetto, S.; Cauda, R.; De Luca, A. Usefulness of Monitoring HIV Drug Resistance and Adherence in Individuals Failing Highly Active Antiretroviral Therapy: A Randomized Study (ARGENTA). AIDS 2002, 16, 369–379. [Google Scholar] [CrossRef]
- Meynard, J.-L.; Vray, M.; Morand-Joubert, L.; Race, E.; Descamps, D.; Peytavin, G.; Matheron, S.; Lamotte, C.; Guiramand, S.; Costagliola, D.; et al. Phenotypic or Genotypic Resistance Testing for Choosing Antiretroviral Therapy after Treatment Failure: A Randomized Trial. AIDS 2002, 16, 727–736. [Google Scholar] [CrossRef]
- Koullias, Y.; Sax, P.E.; Fields, N.F.; Walensky, R.P.; Hyle, E.P. Should We Be Testing for Baseline Integrase Resistance in Patients Newly Diagnosed with Human Immunodeficiency Virus? Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2017, 65, 1274–1281. [Google Scholar] [CrossRef] [Green Version]
- Günthard, H.F.; Calvez, V.; Paredes, R.; Pillay, D.; Shafer, R.W.; Wensing, A.M.; Jacobsen, D.M.; Richman, D.D. Human Immunodeficiency Virus Drug Resistance: 2018 Recommendations of the International Antiviral Society-USA Panel. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2019, 68, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV; NIH Office of AIDS Research Advisory Council (OARAC): Bethesda, MD, USA, 2018. [Google Scholar]
- Gachogo, R.W.; Mwai, D.N.; Onyambu, F.G. Cost Analysis of Implementing HIV Drug Resistance Testing in Kenya: A Case Study of a Service Delivery Site at a Tertiary Level Hospital in Kenya. F1000Research 2020, 9, 793. [Google Scholar] [CrossRef]
- Inzaule, S.C.; Ondoa, P.; Peter, T.; Mugyenyi, P.N.; Stevens, W.S.; de Wit, T.F.R.; Hamers, R.L. Affordable HIV Drug-Resistance Testing for Monitoring of Antiretroviral Therapy in Sub-Saharan Africa. Lancet Infect. Dis. 2016, 16, e267–e275. [Google Scholar] [CrossRef] [PubMed]
- Chrysostomou, A.C.; Topcu, C.; Stylianou, D.C.; Hezka, J.; Kostrikis, L.G. Development of a New Comprehensive HIV-1 Genotypic Drug Resistance Assay for All Commercially Available Reverse Transcriptase, Protease and Integrase Inhibitors in Patients Infected with Group M HIV-1 Strains. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2020, 81, 104243. [Google Scholar] [CrossRef] [PubMed]
- Manyana, S.; Gounder, L.; Pillay, M.; Manasa, J.; Naidoo, K.; Chimukangara, B. HIV-1 Drug Resistance Genotyping in Resource Limited Settings: Current and Future Perspectives in Sequencing Technologies. Viruses 2021, 13, 1125. [Google Scholar] [CrossRef]
- Devereux, H.L.; Youle, M.; Johnson, M.A.; Loveday, C. Rapid Decline in Detectability of HIV-1 Drug Resistance Mutations after Stopping Therapy. AIDS 1999, 13, F123–F127. [Google Scholar] [CrossRef] [PubMed]
- Pujades-Rodríguez, M.; Balkan, S.; Arnould, L.; Brinkhof, M.A.W.; Calmy, A. AIDS Working Group of MSF, for the Treatment Failure and Mortality Factors in Patients Receiving Second-Line HIV Therapy in Resource-Limited Countries. JAMA 2010, 304, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Eholié, S.-P.; Aoussi, F.E.; Ouattara, I.S.; Bissagnéné, E.; Anglaret, X. HIV Treatment and Care in Resource-Constrained Environments: Challenges for the next Decade. J. Int. AIDS Soc. 2012, 15, 17334. [Google Scholar] [CrossRef]
- Eron, J.J.; Clotet, B.; Durant, J.; Katlama, C.; Kumar, P.; Lazzarin, A.; Poizot-Martin, I.; Richmond, G.; Soriano, V.; Ait-Khaled, M.; et al. Safety and Efficacy of Dolutegravir in Treatment-Experienced Subjects with Raltegravir-Resistant HIV Type 1 Infection: 24-Week Results of the VIKING Study. J. Infect. Dis. 2013, 207, 740–748. [Google Scholar] [CrossRef]
- Grinsztejn, B.; Hughes, M.; Ritz, J.; Salata, R.; Mugyenyi, P.; Hogg, E.; Wieclaw, L.; Gross, R.; Godfrey, C.; Kumarasamy, N.; et al. Results of ACTG A5288: A Strategy Study in RLS for 3rd-Line ART Candidates. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 4–7 March 2018. [Google Scholar]
- Lalezari, J.P.; Henry, K.; O’Hearn, M.; Montaner, J.S.G.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J.J.; et al. Enfuvirtide, an HIV-1 Fusion Inhibitor, for Drug-Resistant HIV Infection in North and South America. N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef] [Green Version]
- Gulick, R.M.; Lalezari, J.; Goodrich, J.; Clumeck, N.; DeJesus, E.; Horban, A.; Nadler, J.; Clotet, B.; Karlsson, A.; Wohlfeiler, M.; et al. Maraviroc for Previously Treated Patients with R5 HIV-1 Infection. N. Engl. J. Med. 2008, 359, 1429–1441. [Google Scholar] [CrossRef] [Green Version]
- Emu, B.; Fessel, J.; Schrader, S.; Kumar, P.; Richmond, G.; Win, S.; Weinheimer, S.; Marsolais, C.; Lewis, S. Phase 3 Study of Ibalizumab for Multidrug-Resistant HIV-1. N. Engl. J. Med. 2018, 379, 645–654. [Google Scholar] [CrossRef]
- Hoff, J.; Bani-Sadr, F.; Gassin, M.; Raffi, F. Evaluation of Chronic Hepatitis B Virus (HBV) Infection in Coinfected Patients Receiving Lamivudine as a Component of Anti-Human Immunodeficiency Virus Regimens. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2001, 32, 963–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.hcvguidelines.org/unique-populations/hiv-hcv (accessed on 13 May 2023).
- Benson, C.A.; Brooks, J.T.; Holmes, K.K.; Kaplan, J.E.; Masur, H.; Pau, A. Guidelines for Prevention and Treatment Opportunistic Infections in HIV-Infected Adults and Adolescents: Recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association/Infectious Diseases Society of America; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2009. [Google Scholar]
- Murphy, D.A.; Marelich, W.D.; Hoffman, D.; Steers, W.N. Predictors of Antiretroviral Adherence. AIDS Care 2004, 16, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wilson, T.E.; Adedimeji, A.; Merenstein, D.; Milam, J.; Cohen, J.; Cohen, M.; Golub, E.T. The Impact of Substance Use on Adherence to Antiretroviral Therapy Among HIV-Infected Women in the United States. AIDS Behav. 2018, 22, 896–908. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.L.; Gramling, L.F. Stigma: A Health Barrier for Women with HIV/AIDS. J. Assoc. Nurses AIDS Care JANAC 2004, 15, 30–39. [Google Scholar] [CrossRef]
- Stirratt, M.J.; Remien, R.H.; Smith, A.; Copeland, O.Q.; Dolezal, C.; Krieger, D. The Role of HIV Serostatus Disclosure in Antiretroviral Medication Adherence. AIDS Behav. 2006, 10, 483–493. [Google Scholar] [CrossRef]
- Palmer, N.B.; Salcedo, J.; Miller, A.L.; Winiarski, M.; Arno, P. Psychiatric and Social Barriers to HIV Medication Adherence in a Triply Diagnosed Methadone Population. AIDS Patient Care STDs 2003, 17, 635–644. [Google Scholar] [CrossRef]
- Thompson, M.A.; Mugavero, M.J.; Amico, K.R.; Cargill, V.A.; Chang, L.W.; Gross, R.; Orrell, C.; Altice, F.L.; Bangsberg, D.R.; Bartlett, J.G.; et al. Guidelines for Improving Entry into and Retention in Care and Antiretroviral Adherence for Persons with HIV: Evidence-Based Recommendations from an International Association of Physicians in AIDS Care Panel. Ann. Intern. Med. 2012, 156, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.hiv-druginteractions.org/checker (accessed on 16 May 2023).
- Custodio, J.M.; Yin, X.; Hepner, M.; Ling, K.H.J.; Cheng, A.; Kearney, B.P.; Ramanathan, S. Effect of Food on Rilpivirine/Emtricitabine/Tenofovir Disoproxil Fumarate, an Antiretroviral Single-Tablet Regimen for the Treatment of HIV Infection. J. Clin. Pharmacol. 2014, 54, 378–385. [Google Scholar] [CrossRef]
- Sekar, V.; Kestens, D.; Spinosa-Guzman, S.; De Pauw, M.; De Paepe, E.; Vangeneugden, T.; Lefebvre, E.; Hoetelmans, R.M.W. The Effect of Different Meal Types on the Pharmacokinetics of Darunavir (TMC114)/Ritonavir in HIV-Negative Healthy Volunteers. J. Clin. Pharmacol. 2007, 47, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, M.; Watanabe, K.; Kobayakawa, M.; Mihara, F.; Edamoto, Y.; Teruya, K.; Kikuchi, Y.; Oka, S. Successful Absorption of Antiretroviral Drugs after Gastrojejunal Bypass Surgery Following Failure of Therapy through a Jejunal Tube. Intern. Med. Tokyo Jpn. 2009, 48, 1103–1104. [Google Scholar] [CrossRef]
- Mannheimer, S.B.; Morse, E.; Matts, J.P.; Andrews, L.; Child, C.; Schmetter, B.; Friedland, G.H. Sustained Benefit from a Long-Term Antiretroviral Adherence Intervention. Results of a Large Randomized Clinical Trial. J. Acquir. Immune Defic. Syndr. 2006, 43 (Suppl. S1), S41–S47. [Google Scholar] [CrossRef] [PubMed]
- Gulick, R.M. Investigational Antiretroviral Drugs: What Is Coming Down the Pipeline. Top. Antivir. Med. 2018, 25, 127–132. [Google Scholar]
- Cahn, P.; Fink, V.; Patterson, P. Fostemsavir: A New CD4 Attachment Inhibitor. Curr. Opin. HIV AIDS 2018, 13, 341–345. [Google Scholar] [CrossRef]
- Lataillade, M.; Lalezari, J.P.; Kozal, M.; Aberg, J.A.; Pialoux, G.; Cahn, P.; Thompson, M.; Molina, J.-M.; Moreno, S.; Grinsztejn, B.; et al. Safety and Efficacy of the HIV-1 Attachment Inhibitor Prodrug Fostemsavir in Heavily Treatment-Experienced Individuals: Week 96 Results of the Phase 3 BRIGHTE Study. Lancet HIV 2020, 7, e740–e751. [Google Scholar] [CrossRef]
- Ackerman, P.; Thompson, M.; Molina, J.-M.; Aberg, J.; Cassetti, I.; Kozal, M.; Castagna, A.; Martins, M.; Ramgopal, M.; Sprinz, E.; et al. Long-Term Efficacy and Safety of Fostemsavir among Subgroups of Heavily Treatment-Experienced Adults with HIV-1. AIDS Lond. Engl. 2021, 35, 1061–1072. [Google Scholar] [CrossRef]
- Chang, X.L.; Reed, J.S.; Webb, G.M.; Wu, H.L.; Le, J.; Bateman, K.B.; Greene, J.M.; Pessoa, C.; Waytashek, C.; Weber, W.C.; et al. Suppression of Human and Simian Immunodeficiency Virus Replication with the CCR5-Specific Antibody Leronlimab in Two Species. PLoS Pathog. 2022, 18, e1010396. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, S.; Saladini, F.; Bellocchi, M.C.; Galli, L.; Gagliardini, R.; Gazzola, L.; Francisci, D.; Vichi, F.; Focà, E.; Zazzi, M.; et al. Leronlimab (PRO 140) in Vitro Activity against 4-Class Drug Resistant HIV-1 from Heavily Treatment Experienced Subjects. Pharmacol. Res. 2022, 176, 106064. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Wong, W.-W.; Tsai, H.-C.; Chen, Y.-H.; Kuo, B.-S.; Lynn, S.; Blazkova, J.; Clarridge, K.E.; Su, H.-W.; Lin, C.-Y.; et al. Effect of Anti-CD4 Antibody UB-421 on HIV-1 Rebound after Treatment Interruption. N. Engl. J. Med. 2019, 380, 1535–1545. [Google Scholar] [CrossRef]
- Caskey, M. Broadly Neutralizing Antibodies for the Treatment and Prevention of HIV Infection. Curr. Opin. HIV AIDS 2020, 15, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, P.; Gruell, H.; Nogueira, L.; Pai, J.A.; Butler, A.L.; Millard, K.; Lehmann, C.; Suárez, I.; Oliveira, T.Y.; Lorenzi, J.C.C.; et al. Combination Therapy with Anti-HIV-1 Antibodies Maintains Viral Suppression. Nature 2018, 561, 479–484. [Google Scholar] [CrossRef]
- Ko, S.-Y.; Pegu, A.; Rudicell, R.S.; Yang, Z.; Joyce, M.G.; Chen, X.; Wang, K.; Bao, S.; Kraemer, T.D.; Rath, T.; et al. Enhanced Neonatal Fc Receptor Function Improves Protection against Primate SHIV Infection. Nature 2014, 514, 642–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-On, Y.; Gruell, H.; Schoofs, T.; Pai, J.A.; Nogueira, L.; Butler, A.L.; Millard, K.; Lehmann, C.; Suárez, I.; Oliveira, T.Y.; et al. Safety and Antiviral Activity of Combination HIV-1 Broadly Neutralizing Antibodies in Viremic Individuals. Nat. Med. 2018, 24, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Salie, Z.L.; Kirby, K.A.; Michailidis, E.; Marchand, B.; Singh, K.; Rohan, L.C.; Kodama, E.N.; Mitsuya, H.; Parniak, M.A.; Sarafianos, S.G. Structural Basis of HIV Inhibition by Translocation-Defective RT Inhibitor 4′-Ethynyl-2-Fluoro-2′-Deoxyadenosine (EFdA). Proc. Natl. Acad. Sci. USA 2016, 113, 9274–9279. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/show/NCT05052996 (accessed on 22 May 2023).
- Dvory-Sobol, H.; Shaik, N.; Callebaut, C.; Rhee, M.S. Lenacapavir: A First-in-Class HIV-1 Capsid Inhibitor. Curr. Opin. HIV AIDS 2022, 17, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Margot, N.; Vanderveen, L.; Naik, V.; Ram, R.; Parvangada, P.C.; Martin, R.; Rhee, M.; Callebaut, C. Phenotypic Resistance to Lenacapavir and Monotherapy Efficacy in a Proof-of-Concept Clinical Study. J. Antimicrob. Chemother. 2022, 77, 989–995. [Google Scholar] [CrossRef]
- DeJesus, E.; Harward, S.; Jewell, R.C.; Johnson, M.; Dumont, E.; Wilches, V.; Halliday, F.; Talarico, C.L.; Jeffrey, J.; Gan, J.; et al. A Phase IIa Study Evaluating Safety, Pharmacokinetics, and Antiviral Activity of GSK2838232, a Novel, Second-Generation Maturation Inhibitor, in Participants with Human Immunodeficiency Virus Type 1 Infection. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, 71, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.R.; Fernando, D.; Igwe, S.; McKenzie, L.; Krishnatry, A.S.; Halliday, F.; Zhan, J.; Greene, T.J.; Xu, J.; Ferron-Brady, G.; et al. Phase I Evaluation of the Safety, Tolerability, and Pharmacokinetics of GSK3640254, a next-Generation HIV-1 Maturation Inhibitor. Pharmacol. Res. Perspect. 2020, 8, e00671. [Google Scholar] [CrossRef] [PubMed]
- Spinner, C.D.; Felizarta, F.; Rizzardini, G.; Philibert, P.; Mitha, E.; Domingo, P.; Stephan, C.J.; DeGrosky, M.; Bainbridge, V.; Zhan, J.; et al. Phase IIa Proof-of-Concept Evaluation of the Antiviral Efficacy, Safety, Tolerability, and Pharmacokinetics of the Next-Generation Maturation Inhibitor GSK3640254. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2022, 75, 786–794. [Google Scholar] [CrossRef]
- Saag, M.S.; Gandhi, R.T.; Hoy, J.F.; Landovitz, R.J.; Thompson, M.A.; Sax, P.E.; Smith, D.M.; Benson, C.A.; Buchbinder, S.P.; Del Rio, C.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2020 Recommendations of the International Antiviral Society-USA Panel. JAMA 2020, 324, 1651–1669. [Google Scholar] [CrossRef]
- Overton, E.T.; Richmond, G.; Rizzardini, G.; Jaeger, H.; Orrell, C.; Nagimova, F.; Bredeek, F.; García Deltoro, M.; Swindells, S.; Andrade-Villanueva, J.F.; et al. Long-Acting Cabotegravir and Rilpivirine Dosed Every 2 Months in Adults with HIV-1 Infection (ATLAS-2M), 48-Week Results: A Randomised, Multicentre, Open-Label, Phase 3b, Non-Inferiority Study. Lancet 2020, 396, 1994–2005. [Google Scholar] [CrossRef]
- Hoy, J.; McMahon, J. Is It Time for Injectable Antiretroviral Therapy for HIV? Lancet 2020, 396, 1944–1946. [Google Scholar] [CrossRef]
- Havlir, D.; Gandhi, M. Implementation Challenges for Long-Acting Antivirals as Treatment. Curr. Opin. HIV AIDS 2015, 10, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Marcus, J.L.; Hurley, L.B.; Krakower, D.S.; Alexeeff, S.; Silverberg, M.J.; Volk, J.E. Use of Electronic Health Record Data and Machine Learning to Identify Candidates for HIV Pre-Exposure Prophylaxis: A Modelling Study. Lancet HIV 2019, 6, e688–e695. [Google Scholar] [CrossRef]
- Ahlström, M.G.; Ronit, A.; Omland, L.H.; Vedel, S.; Obel, N. Algorithmic Prediction of HIV Status Using Nation-Wide Electronic Registry Data. EClinicalMedicine 2019, 17, 100203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilkey, M.B.; Marcus, J.L.; Garrell, J.M.; Powell, V.E.; Maloney, K.M.; Krakower, D.S. Using HIV Risk Prediction Tools to Identify Candidates for Pre-Exposure Prophylaxis: Perspectives from Patients and Primary Care Providers. AIDS Patient Care STDs 2019, 33, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Seboka, B.T.; Yehualashet, D.E.; Tesfa, G.A. Artificial Intelligence and Machine Learning Based Prediction of Viral Load and CD4 Status of People Living with HIV (PLWH) on Anti-Retroviral Treatment in Gedeo Zone Public Hospitals. Int. J. Gen. Med. 2023, 16, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Novak, J.; Pathak, P.; Grishina, M.A.; Potemkin, V.A. The Design of Compounds with Desirable Properties–The Anti-HIV Case Study. J. Comput. Chem. 2023, 44, 1016–1030. [Google Scholar] [CrossRef]
- Sarkar, C.; Das, B.; Rawat, V.S.; Wahlang, J.B.; Nongpiur, A.; Tiewsoh, I.; Lyngdoh, N.M.; Das, D.; Bidarolli, M.; Sony, H.T. Artificial Intelligence and Machine Learning Technology Driven Modern Drug Discovery and Development. Int. J. Mol. Sci. 2023, 24, 2026. [Google Scholar] [CrossRef]
- Kumar, S.D.; Singaravelu, G.; Ajithkumar, S.; Murugan, K.; Nicoletti, M.; Benelli, G. Mangrove-Mediated Green Synthesis of Silver Nanoparticles with High HIV-1 Reverse Transcriptase Inhibitory Potential. J. Clust. Sci. 2017, 28, 359–367. [Google Scholar] [CrossRef]
- Maduray, K.; Moodley, R.; Ramdhani, S.; Parboosing, R. The Anti-HIV Activity of Biogenic Silver Nanoparticles Synthesized from Centella Asiatica Extracts. J. Herb. Med. 2022, 35, 100592. [Google Scholar] [CrossRef]
- Priya, M.; Iyer, P.R. Biosynthesis and Optimization of Highly Stable Gold Nanoparticles, Nanoconjugates, Nanodrug Conjugates and Chitosan Nanoconjugates Using Medicinal Plants. Bull. Natl. Res. Cent. 2022, 46, 134. [Google Scholar] [CrossRef]
- Dogra, S.; Sharma, M.D.; Tabassum, S.; Mishra, P.; Bhatt, A.K.; Bhuyar, P. Green Biosynthesis of Silver Nanoparticles (AgNPs) from Vitex Negundo Plant Extract and its Phytochemical Screening and Antimicrobial Assessment Next to Pathogenic Microbes. J. Microbiol. Biotechnol. Food Sci. 2023, 12, 1–7. [Google Scholar]
- Ying, S.; Guan, Z.; Ofoegbu, P.C.; Clubb, P.; Rico, C.; He, F.; Hong, J. Green Synthesis of Nanoparticles: Current Developments and Limitations. Environ. Technol. Innov. 2022, 26, 102336. [Google Scholar] [CrossRef]
- Parveen, K.; Banse, V.; Ledwani, L. Green Synthesis of Nanoparticles: Their Advantages and Disadvantages; AIP Publishing LLC: Melville, NY, USA, 2016; Volume 1724, p. 020048. [Google Scholar]



{kind=link}
{kind=link}
| Class of ART | Drug | Commercial Name | Company | Mutations | References | |
|---|---|---|---|---|---|---|
| Nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) | Abacavir (ABC) | Ziagen® | GlaxoSmithKline, Brentford, Middlesex, UK. | K65R, L74V, Y115F, M184V/I, M41L, D67N, T215Y, K219N/Q, A62V, K70R, V75I, F116Y, Q151M, L210W…etc. | [19,20,21,22] | |
| Emtricitabine (FTC) | Emtriva® | Gilead Sciences, Lakeside Drive Foster City, California, USA. | M184V/I, K65R, T69, Q151M, K43Q/N, E203K, H208Y, D218E, K223Q/E, L228H/R…etc. | |||
| Lamivudine (3TC) | Epivir® | GlaxoSmithKline, Brentford, Middlesex, UK. | M184V/I, E44D, V1181, M41L, D76N, M184V, T215Y, K219N, T215F, K70R, K65R, K219Q, L210W, A62V, L74V…etc. | |||
| Stavudine (d4T) | Zerit® | Bristol Myers-Squibb, Princeton Pike, USA. | M41L, M184V, T215Y, A62V, D67N, K70R, V75I, F116Y, Q151M. K219Q…etc. | |||
| Tenofovir disoproxil fumarate (TDF) | Viread® | Gilead Sciences Lakeside Drive Foster City, California, USA. | K65R, M41L, L210W, L74V, K70E/G/Q/T/N, S68G…etc. | |||
| Tenofovir alafenamide (TAF) | Vemlidy® | Gilead Sciences Lakeside Drive Foster City, California, USA. | ||||
| Didanosine (ddI) | Videx EC® | Bristol Myers-Squibb, Princeton Pike, USA. | L74V, A62V, D67N, K70R, V75L, F116Y, Q151M, K219Q, M41L, M184V, L210W, T215Y, T215F…etc. | |||
| Zidovudine (AZT) | Retrovir® | GlaxoSmithKline, Brentford, Middlesex, UK. | K70R, T215T/F, M41L, K65R, D76N, M184V, T215Y, K219N, T215F…etc. | |||
| Non-nucleoside and nucleoside/nucleotide reverse transcriptase inhibitors (NNRTIs) | Doravirine (DOR) | Pifeltro® | Merck & Co, New Jersey, USA. | A98G, L100I, K103N, V106A, V108I, Y188L, G190S, P225H, V106A, F227L, M230L, L234I, and Y318F | [22,23,24] | |
| Efavirenz (EFV) | Sustiva® | Mylan, Pennsylvania, USA. | K103N, N348I, Y318F, K238N/T, L234I, Y232H, F227L, P225H, G190A/E/Q, Y188L and Y181C | |||
| Etravirine (ETR) | Intelence® | Janssen, Beerse, Belgium. | V90I, A98G, L100I/V, K101E/H/P, V106A/I/M, L234I, E138A/G/K/Q, V179D/E/F/I/L/M/T, Y181C/I/S/V, Y188C/H/L, G190A/C/E/Q/S/T/V, P225H, F227C, M230L, and K238 N/T | |||
| Rilpivirine (RPV) | Edurant® | Tibotec, Mechelen, Belgium. | V90I, L100I, K101E/P/T, V106A/I, V108I, E138A/G/K/Q/R, V179F/I/L, Y181C/I/V, Y188I, G190E, H221Y, F227C/L, and M230I/L | |||
| Delavirdine (DLV) | Rescriptor® | ViiV Healthcare, Brentford Middlesex, UK. | P236L, K103N, Y181C and Y318F | |||
| Dapivirine (DPV) | - | Janssen Therapeutics, Beerse, Belgium. | L100I and K103N | |||
| Nevirapine (NVP) | Viramune® | Boehringer Ingelheim Pharmaceuticals, Ingelheim am Rhein, Germany. | Y181C, V106A, N348I, P236L, L234I, Y232H, G190A/E/Q, M230L, F227L, K103N, P225H, Y188L, Y181C/I/V…etc. | |||
| Protease inhibitors (PIs) | Atazanavir (ATV) | Reyataz® | Bristol Myers Squibb Co. Princeton Pike, USA. | 32I, 33F, 46IL, 47V, 48VM, 50L, 54VTALM, 82ATFS, 84V, 88S, 90M…etc. | [22,25,26] | |
| Darunavir (DRV) | Prezista® | AbbVie Inc, Chicago, USA. | 32I, 33F, 47VA, 50V, 54LM, 76V, 82F, 84V…etc. | |||
| Fosamprenavir (FPV) | Lexiva® | GlaxoSmithKline plc, Brentford, Middlesex, UK. | 32I, 33F, 46IL, 47VA, 50V, 54VTALM, 76V, 82ATFS, 84V, 90M…etc. | |||
| Ritonavir (RTV) | Norvir® | Abbott Laboratories, Illinois, USA. | 10I, I54V, L63P, L76V, A71V, V82A/F, I84V K14R, K20I, E34Q, I47V, I54M, K55R, T74P and I84V | |||
| Saquinavir (SQV) | Invirase® | F. Hoffmann La Roche Ltd. Basel, Switzerland. | 48VM, 54VTALM, 82AT, 84V, 88S, 90M…etc. | |||
| Indinavir (IDV) | Crixivan® | Merck & Co, Inc. New Jersey, USA. | 32I, 46IL, 47V, 54VTALM, 76V, 82ATFS, 84V, 88S, 90M…etc. | |||
| Lopinavir (LPV) | Kaletra® | Abbott Laboratories, Illinois, USA. | 32I, 33F, 46IL, 47VA, 48VM, 50V, 54VTALM, 76V, 82ATFS, 84V, 90M…etc. | |||
| Nelfinavir (NFV) | Viracept® | Agouron Pharmaceuticals, San Diego, California, USA. (Pfizer) | 30N, 33F, 46IL, 47V, 48VM, 54VTALM, 82ATFS, 84V, 88DS, 90M…etc. | |||
| Tipranavir (TPV) | Aptivus® | Boehringer Ingelheim GmbH. Ingelheim am Rhein, Germany. | 32I, 33F, 46IL, 47VA, 54VAM, 82TL, 84V…etc. | |||
| Integrase strand transfer inhibitors (INSTIs) | Dolutegravir (DTG) | Tivicay® | ViiV Healthcare Brentford Middlesex, UK. | E92Q, N155H, G149A, S147G, G118R, S153F/Y, G193E, M50I, R263K, Q148H/K/R…etc. | [22,27,28,29,30] | |
| Raltegravir (RAL) | Isentress® | Merck & Co., Inc. New Jersey, USA. | E92Q, S153Y/F, Q148H, N155H, E157Q, Y143H/R/C, S147G, Q148/H/R/K, L74M, Q95K/R, T97A, E138A/K, G140A/S, V151I, G163R, H183P, Y226C/D/F/H, S230R, D232N…etc. | |||
| Elvitegravir (EVG) | Vitekta® | Gilead Sciences, Lakeside Drive Foster City, California, USA. | T66A/I, E92G/Q, S147G, R263K, Q148R, E157Q, N155H, S153Y/F, S147G, Q148H/K/R, V151I…etc. | |||
| Cabotegravir (CAB) | Vocabria® and Apretude® | Janssen Pharmaceutical Companies (Beerse, Belgium) and ViiV Healthcare (Brentford Middlesex, UK) | H51Y, T66A/I/K, G149A, L74M/I/F, S153Y/F, N155H, G140S, Q148H, T97A, G118R, F121C, Q148H/K/R E138K/A/T…etc. | |||
| Bictegravir (BIC) | Biktarvy® | Gilead Sciences, Lakeside Drive Foster City, California, USA. | R263K, M50I, R263K, E92Q, S153Y/F, Y143R, N155H, G149A, Q148H/K/R, Q148R…etc. | |||
| Entry inhibitors | ARVs that block viral entry (CCR5 receptors antagonists) | Maraviroc (MVC) | Selzentry® | Pfizer, New York, USA. | G11R, P13R, I408A, A316T, I323V, A319S, A25K, I315S, I317S, V169M, N192K, L317W, D462N, N463T, S464T, N465D, L820I, I829V, Y837C…etc. | [31,32,33,34,35] |
| ARVs that inhibit HIV-1 virus fusion | Enfuvirtide (T-20) | Fuzeon® | Hoffmann-La Roche, Basel, Switzerland. | A30V, L33V, L34M, G36D/E/S/V, I37V, V38A/E, Q39H/R, Q40H, N42T/D, N43D, L44M, L45M, R46M, L54M, N140I, T18A, Q40H, L45M, T268A, N126K, E137K, S138A…etc | [26,36] | |
| Post-Attachment Inhibitors | Ibalizumab (IBA) | Trogarzo® | TaiMed Biologics Taipei, Taiwan. | [37,38,39] | ||
| Pharmacokinetic Enhancers | Cobicistat (COBI) | Tybost® | Gilead Sciences, Lakeside Drive Foster City, California, USA. | Not applicable | [15] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foka, F.E.T.; Mufhandu, H.T. Current ARTs, Virologic Failure, and Implications for AIDS Management: A Systematic Review. Viruses 2023, 15, 1732. https://doi.org/10.3390/v15081732
Foka FET, Mufhandu HT. Current ARTs, Virologic Failure, and Implications for AIDS Management: A Systematic Review. Viruses. 2023; 15(8):1732. https://doi.org/10.3390/v15081732
Chicago/Turabian StyleFoka, Frank Eric Tatsing, and Hazel Tumelo Mufhandu. 2023. "Current ARTs, Virologic Failure, and Implications for AIDS Management: A Systematic Review" Viruses 15, no. 8: 1732. https://doi.org/10.3390/v15081732