1. Introduction
ΦGT1 is a lytic podovirus of
Sulfitobacter. Both the host and phage were isolated from a tidal flat and sequenced [
1].
Sulfitobacter is a genus in the marine family
Roseobacteraceae of the order
Rhodobacterales of alphaproteobacteria [
2]. There are relatively few characterized phages of cultured alphaproteobacteria despite metagenomics data indicating that phages of alphaproteobacteria are highly abundant in the world’s oceans [
3]. Observations from Hwang et al. [
1] and its associated GenBank file (MT584811) include that the closest Blast matches are to three temperate podoviruses of
Sulfitobacter marinus [
4], including similarity in frame organization of both structural and nonstructural gene modules. However, the level of similarity in the structural genes is much stronger. The large terminase belongs to pfam04466. Pfam04466 is a subset of the terminases called P22-like [
5]. The tubular tail B (tubeB) protein is distantly P22-like. In addition, there is a weak similarity to P22 ejection proteins (analogous to T7 internal virion proteins). These observations suggest that P22 may be a useful prototype for further comparative studies. However, the P22 matches are far away, requiring profile or Hidden Markov Matrix (HMM) methodology for detection.
Sequenced phages that match by BlastP or Psi-Blast more closely than P22 and which we will place into phylogenetic context are as follows. They are a mixture of lytic and temperate phages and are scattered across a broad host taxonomic range. For most structural genes, the next closest match beyond the
Sulfitobacter phages is EPV2, also known as EBPR2 [
6]. This is a metagenomic construct from a paper mostly about EPV1, which is from a betaproteobacterial host. However, the closest Blast matches of the nonstructural genes of EPV2 (including an integrase) are mostly found in chromosomes of
Roseobacteraceae or other alphaproteobacteria, not betaproteobacteria. So EPV2 probably represents an alphaproteobacterial phage of
Roseobacteraceae or an ancestral bacterium thereof. EPV2 is the only other currently sequenced representative of the ΦGT1 lineage after its entry into
Rhodobacterales, apparently by ~0.35 Gya. Beyond EPV2 there are Blast matches to the lytic
Brucella phages [
7], also known as Perisiviruses with prototype phage Pr; or to temperate
Sinorhizobium phage ΦM5 [
8]. These both inhabit
Rhizobiales (aka.
Hypomicrobiales), which is a different alphaproteobacterial order than the one in which ΦGT1 itself resides. More distant matches are found to a large collection of lytic gammaproteobacterial phages, including
Pseudomonas phage LUZ24 [
9], Enterobacteria phage phiEco32 [
10],
Salmonella phage 7–11 [
11], and
Vibrio phage VPp1 [
12], or to temperate deltaproteobacterial phage Mx8 [
13], and temperate phage 45A6 of
Rhodobacterales [
14]. The relationship of ΦM5 to LUZ24 has been previously noted [
8]. Only virion structural genes exhibit this matching pattern, and those that match ΦM5 and the gammaproteobacterial group do not match the Mx8,
Brucella phage, 45A6 cluster, and vice versa. To complement this matching pattern of few and distant matches, sequences from numerous prospective prophages from the expanding library of sequenced bacterial genomes were used. Unlike the sparse representation of the ΦGT1 lineage in
Rhodobacterales, there appears to have been vigorous propagation of ΦGT1-like temperate phages in
Rhizobiales.
The most common way of conducting comparative characterization of phages is to cluster them by the number of matches found by BlastP or other similarity detection programs. The International Committee on Taxonomy of Viruses (ICTV) publishes a formal taxonomy mainly based on this concept. The prior concept of clustering by tail morphotypes into families [
15]—in this case, podoviruses—is currently being discarded by ICTV [
16]. The Blast-based methodology is unable to deal with highly divergent relationships (such as the relationship of ΦGT1 to the closest well-characterized prototype, P22). The disparate phages mentioned above were all previously classified in
Podoviridae, but are now either unclassified or scattered into numerous small disjoint taxa. ICTV has been proposing alternative family names for some clusters of lytic phages. The process of building a phage taxonomy tends to be confounded by recombinational gene reassortment among phages over time. A recent study [
17] demonstrated that temperate phages exhibit about 10-fold more recombinational reassortment than purely lytic phage families. Correspondingly, the coalescing of temperate phages into new ICTV families has been slow compared to lytic phages. Of the phages mentioned above, the gammaproteobacterial phages are a lytic group that will probably eventually qualify as an ICTV family. It is currently splintered into
Bruynogheviruses,
Krylovviruses,
Bjornviruses,
Vicosaviruses, and
Kuraviruses (at least). We will show that the pathway linking ΦGT1 to the gammaproteobacterial group is through a large population of recombinogenic alphaproteobacterial temperate phages, known only through bacterial genome sequencing and unlikely to appear in the ICTV hierarchy any time soon.
We approached this problem through timetrees rather than a clustering approach. The deepest homology relationships that could be detected with HMM comparisons were organized into time-scaled phylogenetic gene trees for those genes that were amenable. The scaling was founded on a global large terminase subunit alignment [
18] which produces a timetree scaled by rooting at the origin of life on Earth [
19]. The time scale was then transferred to other phage genes through whatever segments of congruence were available. In this mode of thinking, the key elements are ancestors, which are definable as having existed at some point in time and having sent forward some population of descendants. In systematics, the analog of the ancestor is a higher-ranking taxon. In the current viral taxonomy, the higher-ranking taxonomy is based on the Baltimore classification, which was not constructed to reflect common ancestry. This creates a blind spot concerning how old the ancestors are to the phage clusters that do have common ancestry, and what is the history relating those ancestors to each other. With timetrees extrapolated into that blind spot, we seek to provide an objective matrix upon which to build models about the descent of phage gene modules and genomes.
A comparison of our previous analysis of
Pseudomonas phage ΦRIO-1 [
19] with the subsequent ICTV treatment illustrates the difference in perspective. Many of the ΦRIO-1 related phages have been grouped by ICTV into the family
Zobellviridae based on the common content of numerous genes. The common ancestor of
Zobellviridae tracks to ~1.5 Gya. The new ICTV families, where they have been created, generally correspond to ancestors in the 1.0–1.5 Gya range, so we think of that as an upper horizon for the ICTV system for correlating with common ancestry. For ΦRIO-1, deeper relationships were explored, working with only the easiest proteins for making deep trees (large terminase, portal, and tubeB). Those trees coalesced the ΦRIO-1 phages with other extended podoviral families of T7-like (
Autographiviridae), P22-like (
Lederbergviruses), and N4-like phages (
Schitoviridae) in the range of 2.7–3.2 Gya. Above 1.5 and below 3.2 Gya, the similarity is difficult to detect, and information is therefore very sparse. The ΦRIO-1 study found that ΦRIO-1 had points of distant similarity to T7, and less so to P22, while N4 is dissimilar in the qualitative sense of not having a tubeB protein at all.
In this study, the most obvious distant similarity of ΦGT1 to a well-characterized prototype is to P22. We will use even more aggressive similarity detection to explore how consistently ΦGT1 components are related to an ancient P22-like ancestor, and if not, how the descent of ΦGT1 genes is mixed from different ancient ancestors. Working in this upper time range requires a different computational methodology than the typical ClustalW to tree program pipeline. The ΦRIO-1 study used recursive application of HMM methods to add ever more divergent clades to alignment, and used posterior scoring by HHpred in HMM-to-HMM comparisons to judge whether deeply diverged clades could be considered validly aligned, and, if so, in which segments. This practice is updated in this work at the point where alignments produced by other methods fail validation by HHpred HMM-to-HMM comparison. In that time zone, alignments of divergent clades were fused by enforcing the HHpred HMM-to-HMM alignment, thus clustering genes even more deeply into evolutionary history. ICTV is also incorporating HMM-to-HMM comparisons [
20]. We differ from their approach by making a targeted effort to make robust broad-based alignments and HMMs for the proteins specific to the podo- (or myo- or sipho-) viral morphology groups, rigorously excluding recombination junctions from the sequences presented to the treemaking process, and evaluating the depth of common ancestors using the timetree paradigm.
With the abandonment of the morphological-tailed phage families, there are now no official names with which to describe clusters or hypothetical clusters of phages arising in the first 2 billion years of life. It is clear from the analysis of Hardies et al. [
19] of the functional and sequence similarity of T7 and P22 tubeB protein that the podoviral ejectosomal mechanism was in existence by ~3.2 Gya. In a taxonomy designed to describe the full history of tailed phages, taxa dating to this period where bacteria form a small number of phyla would be phage phyla, and ejectosomal podoviruses would be one such phylum. From this molecular phylogeny perspective, “ejectosomal” podoviruses means all viruses that descended and retained the requisite gene set from the ancestor that invented the ejectosomal mechanism in the first place. We specify ejectosomal podoviruses rather than just podoviruses as the relevant universe of viruses to consider for this study to exclude phi29 and all related nonejectosomal podoviruses. These have different ancestry and functionality of their tail genes. Prospective subdivisions of the ejectosomal podoviruses in the 1.5 to 3.2 Gya range would be provisional podoviral classes or subclasses. These definitions can merge relatively gracefully with the ICTV system that becomes relevant below 1.5 Gya. It is helpful to have terms to mark these two distinctive epochs of time, so we will call 0–1.5 Gya the Blastoscene, and 1.5–3.8 Gya the Tailoscene. To avoid proposing and arguing about yet another naming system, we will name our ancestors of interest by the time-honored practice of naming one or more well-known descendant prototypical phage, but clarify by adding the estimated time of the common ancestor.
Another distinction of the timetree approach is that it allows a comparison of the timing of phage gene lineage splits with the timing of taxon differentiation in the host phylogeny. There is much discussion in the literature about phage mosaicism. Phages swap genes with other phages. There is a tendency to apply the term “horizontal transfer” to every gene newly acquired by a phage. For example, Mavrich and Hatfull [
17] follow this convention. To us, a transfer from a source in a far away host taxon has different selective and mechanistic implications than picking up a gene from a phage descending in the same host range. With a time scale, we can distinguish those processes. We will use the term “reassortment” to describe the common shuttling about of genes among phages in the same host range. We will reserve the term “horizontal transfer” for a situation where the phage gene had to at some point move horizontally with respect to the host phylogeny.
This paper will focus on the descent of the ΦGT1 core structure genes. One peculiarity of the ΦGT1 genome is that HMM searching reveals two adjacent divergent homologs of the gene for the protein called tubular tail A (tubeA) in T7 and head completion protein, gene 4, in P22. This is the protein in the tail to which the tail fibers attach. We find that an ancestor with the ΦGT1-like dual tubeA structure gave rise to a large temperate phage family in Rhizobiales. The expansion of the dual tubeA family was tracked in Rhizobiales, along with several horizontal transferants cast off by it, including the path to ΦGT1. The origin of this dual tubeA structure was attributed to a recombination that occurred ~0.7 Gya between two more ancient lineages that both had the typical P22 structure module gene order. One ancestor descended from a LUZ24/phiEco32/alphaproteobacterial common ancestor ~1.56 Gya and donated the head structure genes and one copy of tubeA. The other ancestor descended from a Mx8/Brucella phage common ancestor ~1.25 Gya and donated the other copy of tubeA and the remaining tail module. The structural gene modules of both the LUZ24 and Mx8 ancestors have common ancestry with P22 >> 2.0 Gya.
2. Materials and Methods
2.1. Related Prophages
The search for related intact prophages started by searching completely sequenced bacterial chromosomes for close homologs to the ΦGT1 tubeB protein. TubeB was keyed upon to focus on podoviruses. To find an uninserted version of the same chromosome, the DnaE gene in that chromosome was used to key a BlastP search to find a collection of sequenced strains that were very closely related to the one containing the prospective prophage. A region of +/− 100 kb from the query chromosome was used to key a BlastN search of the DNA from those closely related chromosomes reported in the “-outmt 4” aligned format, and the gene annotations from the query sequence were transferred to the alignment. An ideal result would be a gap of ~40 kb starting in one or more closely related genomes within a tRNA gene adjacent to a gene labeled as an integrase in the query chromosome such that the tRNA gene was partially duplicated during the phage insertion. This pattern makes use of the frequent occurrence of phage integrases that use a host tRNA gene as the host attachment site and a partial version of the tRNA gene as the phage attachment site [
21]. Prospective intact prophages were then examined for a full complement of core genes and a lack of genes disrupted by nonsense codons or frameshifts.
2.2. Homolog Candidates and Alignment with the UCSC Sequence Alignment and Modeling System
Alignments were made and extended to highly divergent sequences similar to the method described [
19]. Typically, sets of prospective homologs were found by Psi-Blast searches starting from the ΦGT1 gene or from candidates for homologs in distant phages suggested by similarity in gene size and genomic position, or other criteria. Psi-Blast [
22] was installed locally from the NCBI Toolkit so that databases other than the standard NCBI nr database could be easily searched, for example, nr fused to the metagenomic env_nr, or databases reduced to just tailed phages, or just podoviruses, or just the proteomes of selected individual phages. Psi-Blast iterations were run until either convergence or the accumulation of obvious nonhomologous sequences. The proposed homologs will be screened for significant similarity by a sensitive HMM-based method in the next step, allowing this initial step to be optimized for high sensitivity with less concern about the inclusion of false positives. Homolog candidates were then screened and aligned by the UCSC Sequence Alignment and Modeling system (SAM ver. 3.5) [
23,
24] obtained from Kevin Karplus and Richard Hughey [
https://users.soe.ucsc.edu/~karplus/projects-compbio-html/sam2src/ (accessed on 16 December 2022)]. Although we have also used more common alignment systems, SAM has proven particularly useful in its property of iteratively screening a prospective homolog set to include only sequences with statistical similarity to the developing HMM, and then incorporating those sequences and reoptimizing the HMM and its underlying alignment for the next iteration.
2.3. Alignment Validation
After preliminary tree construction using neighbor-joining in PAUP [
25], or MrBayes, sequences forming the most divergent clades were realigned independently and compared to each other by HHpred [HHsuite ver. 2.0.15 installed locally; most recent version obtainable at
https://github.com/soedinglab/hh-suite (accessed on 27 March 2022)] [
26]. Models were built including secondary structure prediction with the included addss utility. Models were calibrated against the scop70_1.72 HHpred model library and searched against each other one to one by hhsearch with one designated as the query and the other designated as the database. This method produces posterior residue alignment confidence scores. Strings of confidence scores of mainly 7, 8, or 9 were taken to indicate regions of confident alignment. The HHpred alignment was compared to the SAM alignment to confirm that the two methods were in agreement in the confidently aligned segments. If there was no confidently aligned segment, the divergent clade was removed from the analysis. If removal of the non-confidently aligned segments did not alter the tree, they were left in place, since they presumably improve confidence in the lower parts of the tree. If HHpred found a confident alignment and SAM failed to agree, subfamily alignments were fused by forcing a merged alignment to adhere to the HHpred result. Whenever this happened, it is indicated in the legend to the
supplementary tree figure. In principle, the program Clustal Omega conducts this same operation. However, in practice, we found processing alignments by SAM to be more successful in sweeping gaps out of regions with predicted secondary structures.
2.4. Availability of Alignments and HMMs
Alignments and HMMs from this study are available at
https://Stephen-Hardies.github.io (accessed on 10 May 2023). Some of these protein families may eventually appear in Pfam, but in many cases, the criteria used by Pfam contradict the criterion we used for forming the alignment. Pfam prioritizes confident family assignment in a large database search and seeks to limit confusion caused by related families by making separate models for each. We prioritize the HMM as a tool to align distant homologs, which requires all the related subfamilies to be present in the same alignment. If used blindly, say to annotate an entire bacterial genome, these broader models are more prone to raise false positives. However, when used for alignment of genes and treemaking with validation procedures, false positives tend to be much less of a problem. The HMMs provide a direct method to align any collection of related sequences using either hmmeralign or Clustal Omega.
2.5. Treemaking
For the final tree analysis, a small subset of sequences was extracted from the larger alignments made above. Trees were constructed by MrBayes (ver. 3.2.6) [
27] with the BLOSUM replacement matrix as the amino acid prior, a four-category gamma correction, and the independent gamma rates relaxed clock model, also called the “white noise” model [
28], with 4 chains and 1,000,000 generations and 50% reserved for burnin, unless the convergence statistics indicated a need for more generations to achieve convergence. Consensus trees were compiled by the sumt tree utility with the “allcompat” option, and displayed and scaled using Figtree [
http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 8 December 2015)]. Divergence times for host taxa for comparison were from the TimeTree website [
http://timetree.org (accessed on 3 May 2023)] [
29]. For comparison to maximum likelihood, the same alignment was analyzed by MegaX [
30].
2.6. Tree Quality
Several controls were employed to guard against low-quality trees. If subtrees produced from separate domains or of the sequence arbitrarily divided in half did not appear mutually congruent, the tree was rejected, and a search was conducted for the underlying recombinant sequence(s) and recombination points. The tree was then remade without the inclusion of recombination junctions. If addition or subtraction of sequences made the tree incongruent with its former self, the tree was rejected and a search was conducted for the underlying cause, which might be intragenic recombination, substantial poorly aligned segments, or branches or clades evolving at a systematically different rate. The tree was then remade without the problematic segments or sequences.
2.7. Tree Scaling and Congruency
The large terminase tree was made with a selection of far-diverged terminases and the root set at 3.8 Gya—the approximate origin of life on Earth (3.7–4.2) [
31]. For two protein trees to be considered candidates for congruence it was expected that their trees could be aligned with consistent topology so that there were a series of nodes for which the median node height of each fell within ~1.4 times the 95% Height Posterior Density (HPD) of the other tree. The 1.4 multiplier was an effort to account for the joint uncertainty of both trees. Collections of prospectively congruent nodes in the phiEco32/LUZ24 family and ΦGT1 family derived as explained in
Section 3.3 are given in
Table S1A,B. Averages node heights are given excluding any node that appears to fail the congruence test with the others or for which the quality of information is otherwise less than the others. The time scale of each timetree was fine-tuned to maximize agreement with these averages. Candidate trees were first scaled to make the highest of the candidate nodes of equal time to the value from
Table S1A,B. Nodes that failed the congruency test with the reference values were excluded. The deviations of each remaining candidate congruent node from the average of the collection were normalized by 1/4 the 95% HPD, essentially expressing them in numbers of standard deviations. The deviations were then summed, and the time-scale factor was adjusted to make the sum equal to zero. Nodes excluded or additional calibration points added are listed in legends to the
supplementary tree figures.
2.8. Mass Spectrometry
Mass spectrometry was carried out at the National Instrumentation Center for Environmental Management, Seoul National University. RP-nano LC-ESI-MS/MS analysis was conducted using a Thermo Scientific Q Exactive Hybrid Quadrupole-Orbitrap instrument (Thermo Scientific, CA, USA) equipped with Dionex U 3000 RSLCnano HPLC system. Fractions were reconstituted in solvent A (Water/Acetonitrile, 98:2 v/v, 0.1% Formic acid) and then injected into the LC-nano ESI-MS/MS system. Samples were first trapped on an Acclaim PepMap 100 trap column (100 μm × 2 cm, nanoViper C18, 5 μm, 100 Å, Thermo Scientific, part number 164564) and washed for 6 min with 98% solvent A at a flow rate of 4 μL min−1, and then separated on an Acclaim PepMap 100 capillary column (75 μm × 15 cm, nanoViper C18, 3 μm, 100 Å, Thermo Scientific, part number 164568) at a flow rate of 300 nl min−1. The LC gradient was run at 2% to 35% solvent B (80% acetonitrile, 0.1% formic acid) over 30 min, then from 35% to 90% over 10 min, followed by 90% solvent B for 5 min, and finally 5% solvent B for 15 min. The Orbitrap analyzer scanned precursor ions with a mass range of 350–1800 m/z with 70,000 resolution at m/z 200. Xcaliber software (version 3.1) was used to collect MS data and Mascot (Matrix Science, London, UK) was used to search the spectra against a database of predicted ΦGT1 proteins concatenated with the Swiss-Prot database. Mascot results were correlated with X! Tandem with Scaffold (Proteome Software, Inc., OR, USA).
2.9. Ascertainment of Membership in Morphological Families
In support of statements that a lineage is either podovirus-specific or contains a mixture of podo-, sipho-, and myoviruses, a problem has arisen in that NCBI has begun expunging the morphological family names from phage entries. Hence one can no longer research the morphotypes of a collection of protein accessions mapped to a tree by any operation performed on the current NCBI database. As a workaround, we restrict a preliminary survey to phages present in a saved back copy of the NCBI database as of 31 May 2019, before the morphotype information was expunged. The finding was only considered definitive if an associated paper cited the EM examination. The annotation of morphological family types for metagenomic phage sequences was ignored.
2.10. Neighborhood Analysis
To determine if a gene is consistently located in the same gene neighborhood, we performed the following analysis: NCBI was interrogated to find the nucleotide sequence encoding each gene in the tabulated family of the query sequence. Protein sequences defined as features within the nucleotide sequences were retrieved corresponding to a neighborhood defined as some number of genes upstream and downstream. The protein sequences were formed into a library with each entry carrying its original annotation plus its position in the number of genes upstream or downstream relative to the query sequence. The annotation of the sequences in this library was updated with HMM search results for landmark genes in the neighborhood of the original query. The frequency at which homologs of the landmark genes fell within the neighborhood of homologs of the original query could then be directly examined.
2.11. Clade Trees
The trees in
Section 3.8 were designed to reveal how the abundance of the temperate phage families fluctuated in different host taxa and taxa inferred from metagenomic data. For prophages, the analysis was confined to completely sequenced and assembled bacterial chromosomes that were retrieved from NCBI 5/2019 and curated to incorporate a full taxonomic description within each protein definition line and to avoid plasmids. It had 17,457 genomes. This was intended to facilitate counting the number of sequenced chromosomes that yielded the number of prophage homologs found in any taxonomic envelope. The metagenomic collection was retrieved from NCBI 10/2020 with an Entrez query including “uncultured phage” in the definition line. It had 3187 genomes. Search and alignment by Psi-Blast and SAM were conducted as in
Section 2.2 with the inclusion of a blast-formatted version of this metagenomic collection. Clades were defined by preliminary neighbor-joining analysis. Exemplars from each clade were subjected to MrBayes analysis to create the time scale for the clade trees. The final tree construction was as described in the figure legend.
2.12. Replacement/Synonymous Analysis
Protein sequence alignments were converted to nucleotide alignments using pal2nal [
32]. Replacement and synonymous changes were counted and normalized by the number of sites using the DNAStatistics module of the BioPerl Align package.
4. Discussion
4.1. Relationship of ΦGT1 to the Common Ancestral Podovirus
We have shifted our analysis of phage genomes to timetrees to have a more quantitative basis for examining congruence and to more clearly picture the early times during which the gene modules were established. Modules exhibiting coherent descent over the longest periods of time were prioritized for establishing an overall evolutionary framework of the family’s descent. The time scale was set to make the initial radiation of the packaging ATPase system at 3.8 Gya, consistent with the theory that phages were either involved in the invention of DNA and the transition from RNA to DNA world or appeared very soon thereafter [
19]. There is a range of opinions on the timing of the origin of life (reviewed [
31]). However, given that the uncertainty of the reconstructions covers about 1 Gyr at that depth in time, we have picked the 3.8 Gya number as a commonly quoted estimate that is approximately in the middle of the plausible range. The proposition is that this will allow us to place common ancestors in the phage tree into the context of bacterial time trees roughly within the uncertainty intervals shown by the node bars on the trees. The best support for this proposition is found in
Figure S2, in which we used this system to time the mitochondrial endosymbiosis with a phage gene, and found an estimate consistent with the current consensus opinion of the time for that event.
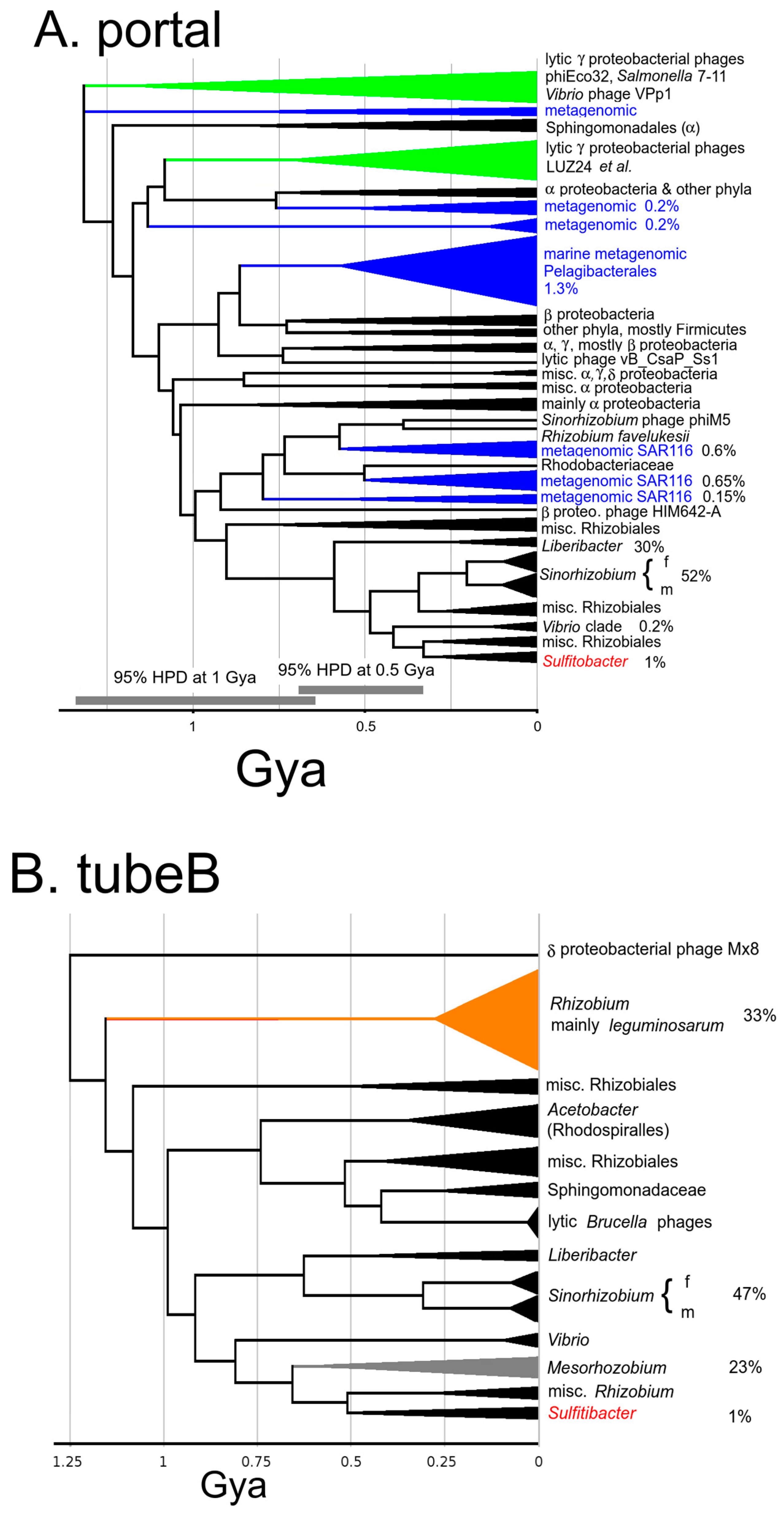
On that time scale, the most anciently established trait of ΦGT1 is that it is a podovirus with an ejectosome. This kind of virus was established by 3.2 Gya and the form has descended maintaining a coherent collection of structural components. This kind of podovirus is most easily recognized at the sequence level by an encoded tubeB protein followed by candidate genes for the ejectosomal proteins themselves. By ~1.5 Gya the ancestors to ΦGT1 have coalesced into two families which each has become mainly committed to a specific structural gene module: the LUZ24/phiEco32 family and the Mx8/Pr family. These two families subsequently recombine to make the ΦGT1-like genome. Both families feature more P22-like than T7-like tubeB proteins by HMM scoring, but by tree analysis diverged in the early Tailoscene soon after or at the same time as the T7/P22 split. Among the other proteins traced into this time zone, the most consistent feature is that the LUZ24/phiEco32 family is highly divergent from the Mx8/Pr family, having split in the early Tailoscene either a little after T7/P22 or as a separate clade from T7+P22. Multiple tubeA lineages were traced to radiation in the early Tailoscene, and have as of yet no representation in any sipho- or myovirus. There may be one or two podovirus-specific portal lineages arising in the early Tailoscene. However, the major head protein appears to be more interchangeable with siphoviruses and myoviruses, and the packaging system is even more so. Thus far, the proposition holds up that to explore ΦGT1 relationships concerning virion structure, assembly, and ejection mechanism to a heavily characterized prototype, P22 would seem to be a promising candidate, although thinking of the LUZ24/phiEco32 lineage, the Mx8/Pr lineage, the T7 lineage, and the P22 lineage as part of unordered ancient radiation is also a good model.
The Mx8/Pr family is not as well populated among sequenced genomes as the LUZ24/phiEco32 family. Although there are numerous
Brucella phages sequenced, they are all nearly identical. Turner et al. [
16] recently published a rationale for removing
Podovirdae from the official phage taxonomy citing as justification that orthologous core genes could not be established between
Lederbergviruses (P22) and
Myxoctoviruses (aka. Mx8), citing a CoreGenes analysis. A relationship between Mx8 and P22 for all the core structural genes is established in this study timing to ancestors ranging from ~2 to 3.5 Gya, with the main ambiguity being whether it is a class of equal depth to P22 of >3 Gya or a subclass of P22 with a major component of core genes related in the 2–3 Gya range. Although we have not included a detailed analysis of the internal virion protein genes here, ΦGT1 IVPB and IVPD find Psi-Blast matches in the syntenic genes of Mx8, leaving little doubt that Mx8 has the classic podoviral structural gene complement. The discrepancy with Turner et al. [
16] can be understood as due to the reliance on computational tools that cannot detect similarity in the requisite genes in the 2+ Gya range.
At ~0.7 Gya the LUZ24/phiEco32 lineage recombined with the Mx8/Pr lineage to create the dual tubeA arrangement of the recombinant ΦGT1 family. This recombination event was described in
Section 3.2 and timed on each tree in the manuscript and in the
Supplemental figures as the node connecting ΦGT1 to the
Sinorhizobium prophages. The ΦGT1 dual tubeA arrangement descends with little apparent revision of the core structural genes. However, there may well be recombination among recent sibling lineages. Those would not be resolvable within the uncertainty of the tree analysis. The nonstructural gene component among the various ΦGT1 clades was essentially replaced within a mere 0.1 Gya (
Section 3.7). Since that degree of churning disrupts establishing congruence with the more coherently descending genes, we do not currently have an applicable method to include them in our analysis.
The junction between the two structural modules is the most unique feature of the ΦGT1-like genome, with two adjacent and highly diverged tubeA genes flanking the junction. That recombination is mapped to a time ~0.7 Gya and has succeeded in spreading into a variety of current host genera. This knowledge can be used to enable a hypothesis-driven approach for further examination of the phage. If one were to choose ΦGT1 for structural studies, for example, one would be armed with the prior knowledge that there is some major structural variation to find, that it involves the tail hub, and that it must be functionally significant to explain the duration and degree of spread of descendants. Since tubeA is a major attachment point for tail fibers and there also appears to be an excess of tail fiber genes in ΦGT1, one would expect to clarify if there is a connection between the unusual tail hub and the number and/or diversity of tail fibers used. One would be alerted that there will either be two rings of the tubeA protein, or one ring of a heterodimer. If there are one or two rings should be clear by even preliminary structural alignment to the ancestral P22 and T7 arrangements. If there was one ring, that would convert the usual 6-fold symmetry of the tail to 3-fold symmetry in approximate 6-fold symmetry. One would be alerted to look for other aspects of 3-fold symmetry imposed on the whole of the tail hub and whether that might impact how the tail fibers transduce the signal through the tubeA ring that the phage is properly positioned for injection. In considering how any altered functionality arose, one could reasonably assume that both ancestors had tail fiber function analogous to T7 or P22 and that the immediate result of the fusion event had to be a functional phage. Recognition of the nature of the dualA arrangement enables other kinds of hypothesis-driven investigation. After being alerted to the possibility of two highly divergent tubeA genes in the same genome, we found another example in the Bcep22-like phages, apparently arising completely independently. Finding this odd recombinant pattern more than once supports the proposition that it has some selective value.
4.2. Relationship of the ΦGT1 Family to the Oceanic Metagenome
Liang et al. [
2] associate
Sulfitobacter with the marine family
Roseobacteraceae. However, although the ΦGT1 genome has multitudinous Psi-Blast matches to metagenomic marine phages, a detailed reconstruction of its history does not include descent as any major phage lineage in
Roseobacteraceae. The Psi-Blast matches reflect shared ancestry with multiple major marine metagenomic lineages as of the time 1–1.5 Gya when the LUZ24 module entered alphaproteobacteria and distributed them into multiple developing orders. The path to ΦGT1 did not go through those marine phage lineages. Instead, it went through
Rhizobiales from which it then transferred to
Roseobacteraceae and made a lineage descending into
Sulfitobacter. Based on the numbers of close Psi-Blast matches in bacterial and metagenomic databases, that lineage appears to be relatively unproductive, barring some sampling bias that is causing it to be underrepresented. In retrospect, the marine metagenome thus far examined is relatively strongly biased to a narrow slice of oligotrophic planktonic hosts. Marine bacteria found in coastal sediments are a very different niche.
4.3. Balance between Horizontal Transfer and Vertical Descent
Incorporation of the many prophages and prophage remnants into the study gives a much more robust accounting of the evolutionary history leading to ΦGT1. This was exploited to explore the balance between the vertical descent of phages from ancestral host taxa versus horizontal transfer across the host range. Since the pivotal review by Hendrix et al. [
40] on horizontal transfer and gene reassortment in phages, mentions of vertical descent in phages have virtually disappeared from the phage literature. We make a distinction between gene reassortment and horizontal transfer across host taxa for the following reason. If phage genes only engaged in recombination within a host taxon but never transferred horizontally among them, then every phage gene would ultimately have the same evolutionary history as its host. There could still be confusion based on recombination between paralogous phage lineages within the same host, but without horizontal transfer between host taxa, phage phylogeny would be a much simpler affair. To consider the balance between horizontal transfer and vertical descent, it is necessary to have some confidence in the timing of phage nodes versus host nodes. Quality controls are offered in
Figures S1 and S2 to document the degree of success and limitations in our current effort to correlate host and phage phylogenies.
Confinement of phages to vertical descent is obvious on a laboratory time scale and is also reflected in the observation that the most recent phage sequence clusters are mostly, although not completely, confined to a host genus. With the greater time resolution provided by timetrees, we see that the origin of the dual tubeA ΦGT1-like structure module early in Rhizobiales is followed by its descendants being concentrated in families and genera of that order. By following the descent of the tubeB gene in that module, we see it passing through the differentiation of families and genera in Rhizobiales before transferring to the sister Rhodobacterales order to descend into ΦGT1. The simplest explanation would be if phage transmission is also mostly vertical on the 0.5 Gyr time scale, although with enough horizontal transfer mixed in to generate ample confusion. After placing the dual tubeA ancestor in Rhizobiales before the generation of the host families, one might expect that all the families and all the genera of Rhizobiales would have descendant ΦGT1-like temperate phage lineages; but they do not. Even after accounting for the differences in the numbers of completely sequenced genomes, the success in vertical transmission is remarkably patchy. One genus was found, Liberibacter, where there are ΦGT1-like copies, but all copies found are full of nonsense codons. So ΦGT1 phages in Liberibacter appear to be in the process of failing. Patchy distribution in vertical descent requires that there be at least soft boundaries to horizontal transfer that develop as bacterial taxa differentiate.
Mixed with a history of patchy vertical descent there are clear examples of horizontal transfer, meaning that the node relating to the phage genes is significantly younger than the node relating to the hosts that they occupy. For example, dual tubeA ΦGT1-like genomes in Rhizobiales and Rhodobacterales are related at ~0.7 Gya. Those host orders diverged by >1.6 Gya according to the TimeTree website. Hence that represents a horizontal transfer when the hosts were about as diverged as bacterial families are today. The dual tubeA module is well represented in Rhizobiales, both in total numbers and numbers of genera and species occupied. However, in Rhodobacterales it is a minor lineage and found only in the family Roseobacteraceae. We consider it most likely, based on a simple mass-action argument that the source of the transfers was the more heavily populated host taxon. The entire core structural module appears to have been transferred intact. Similarly, we found that the dual tubeA genome made a transfer to form a very small clade back in gammaproteobacteria confined to the genus Vibrio. We could find no evidence that any of the other component genes of the phage succeeded in making the transient together with the core genes, either in the homologous prophages or scattered into any other sequenced prophages in the recipient host taxa. That could mean either that only part of the phage genome was transferred, or that only part of the phage genome had sufficient selective value to be retained and amplified after it was transferred.
For horizontal transfers occurring in the last 0.5 Gya, it seems typically true that the recipient side of the transfer is lightly populated. Another example is
Sinorhizobium phage ΦM5. We initially thought that since the LUZ24 donor to make the dual tubeA genome was apparently in early
Rhizobiales that this descendant LUZ24-like phage was another example of vertical descent within
Rhizobiales. However, although ΦM5 is a temperate phage of
Sinorhizobium, we found no relative prophages of it in sequenced
Sinorhizobium genomes, and only one prospective prophage anywhere else in
Rhizobiales. When metagenomic sequence was added, it became very clear that at least the early descent of ΦM5 occurred in the marine bacterial SAR116 taxon (
Section 3.8), which upon examination by Lee et al. [
41] would appear to be a separate alphaproteobacterial order. It would not be surprising if other clean lines of vertical descent were discovered upon collection of additional sequences to have detoured through some remote host taxon.
The earliest horizontal transfers we have characterized are of the LUZ24 module between gamma- and alphaproteobacteria, and of the Mx8 module between delta and alphaproteobacteria. In each case, the transfer is well after the differentiation of the host classes, but we have no evidence at this time to conclude in which host classes the modules originated. Another source of ambiguity about horizontal transfers is that if the transfer is soon after the host differentiation, the relative timing of the nodes will not be different enough to be distinguishable. Since some models would suggest that recently related hosts are much more likely to exchange DNA, that effect could cause the timetrees to gloss over considerable chaos.
One way to clarify the ambiguities of measuring both horizontal transfer and vertical descent is to propose that the load of phage lineages is stationary (meaning an observer at any point in time would see approximately the same load of phage lineages per host taxon). Since there is some horizontal acquisition of phage lineages, a balancing loss of phage lineages is nearly a theoretical necessity. Otherwise, over long evolutionary time, the predicted diversity of phage lineages per taxon would be predicted to continuously increase, possibly even exponentially. Incorporating a balance between lineages gained and lineages lost would avoid those sorts of instabilities and might provide a useful guide for sorting out the observational data.
4.4. Onset of the Host Range Barrier
On the low time zone end of the vertical vs. horizontal transmission problem, we looked at more detail at the
Sinorhizobium “m” vs. “f” lineages. In light of the considerations above, a soft barrier to horizontal transfer does have to at some time arise between diverging host taxa. It would have to be strong enough to explain the frequent observation that one descendant bacterial family or genus vertically inherits a phage lineage, and a sibling host taxon does not. Taking the diversification between
S. meliloti and
S. fredii as an example of a phage vertically transiting a host diversification, we note that the present TimeTree website estimation of the
meliloti/
fredii split is 67 Mya, in good agreement with our best estimate of the differentiation of the “m” and “f” lineages at ~77 Mya, with “m” prophages dominating in
S. meliloti and “f” prophages dominating in
S. fredii (
Section 3.7). The criteria used by taxonomists to declare different bacterial species are many and always in a state of flux, but the agreement between these two numbers suggests that the consensus of criteria about what makes these different species of bacteria has matched the onset of a barrier that would allow the vertically descending phage lineage to differentiate into
meliloti and
fredii branches. However, when the analysis was extended to a collection of 20 “m” and “f” prophages, three instances of a bacterial strain containing one each of them were found. This would seem to suggest that at its inception, the boundary is much leakier than it will eventually become.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




