
Modulation of Reoviral Cytolysis (II): Cellular Stemness
 and
and 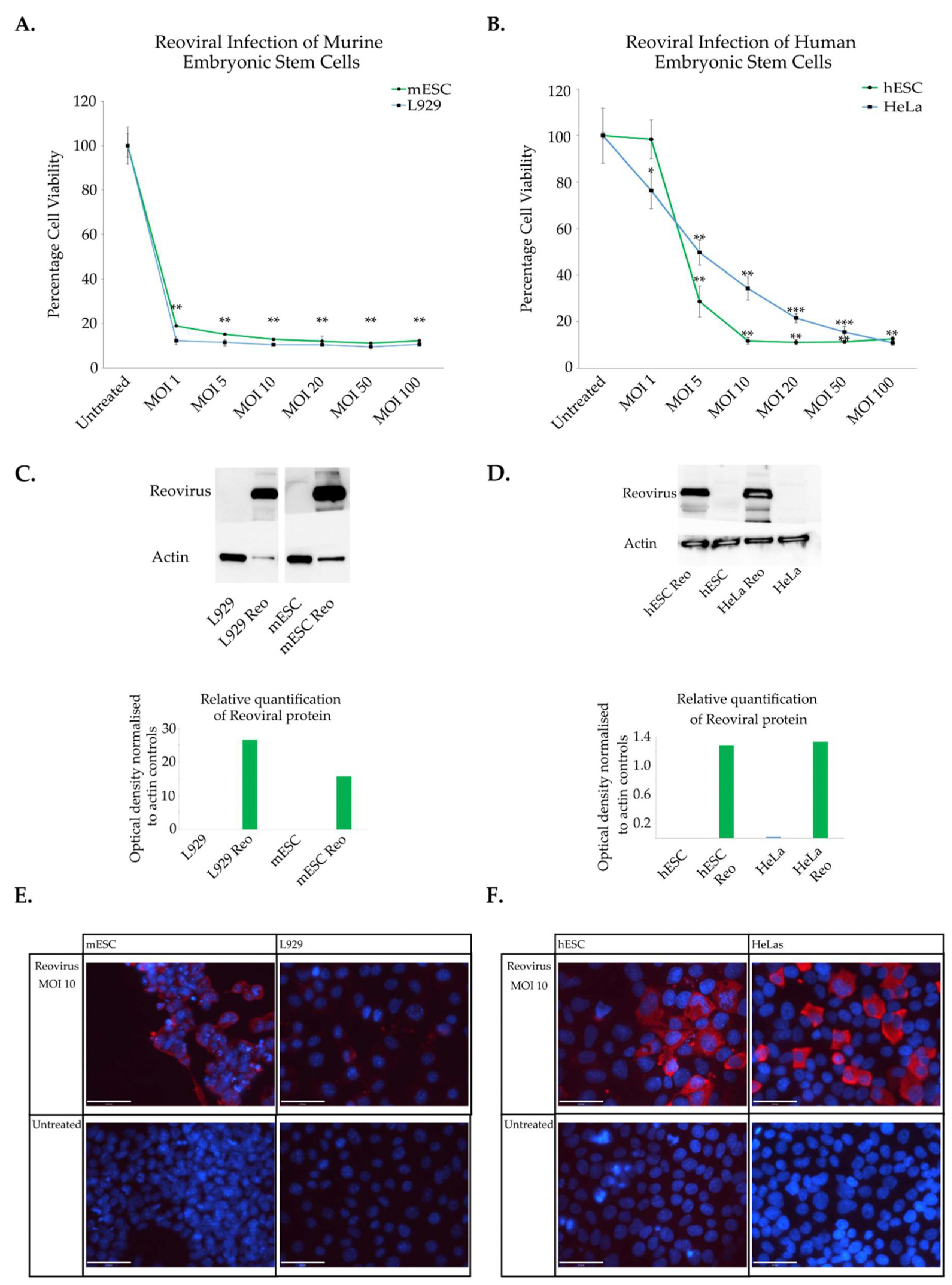{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cancer Cell Lines and Culture Conditions
2.2. Stem Cell Culture Conditions
2.2.1. Murine Cell Lines
2.2.2. Human Stem Cells
2.3. Differentiation Protocols
2.3.1. Murine Cell Lines
2.3.2. Human Cell Lines
2.3.3. Reoviral Infection
2.4. Viability Assays
2.5. Reovirus Protein Synthesis
Immunostaining and Widefield Microscopy
2.6. Western Blots
2.7. Bioinformatic Analysis
2.7.1. Analysis of Reoviral Sensitive vs. Resistant Cell Lines
2.7.2. Analysis of Gene Expression in Tumours
2.8. Statistical Analysis
3. Results
3.1. Characterisation of Reoviral Cytolysis in Embryonic Stem Cells
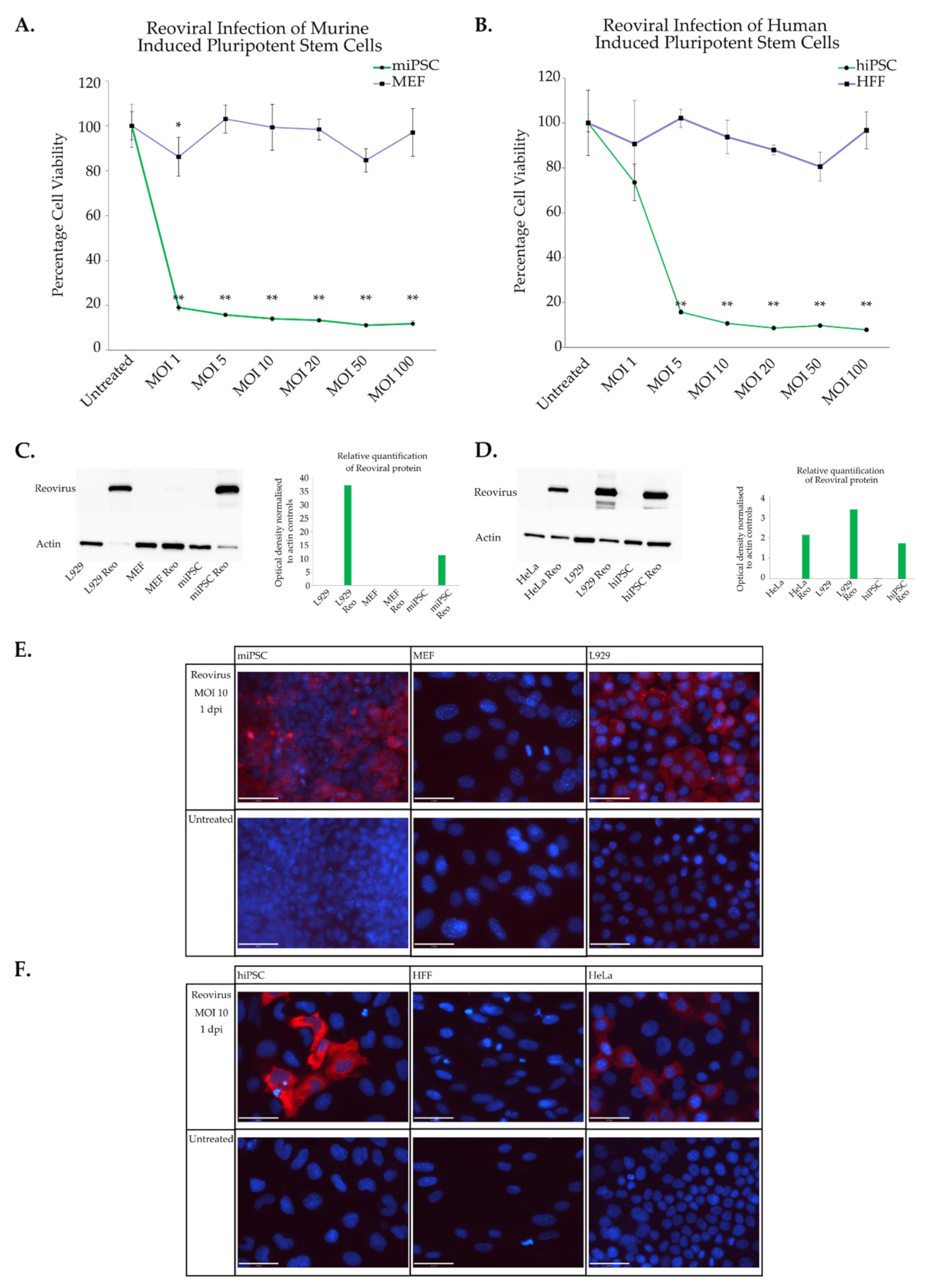
3.2. Characterisation of Reoviral Cytolysis in Induced Pluripotent Stem Cells
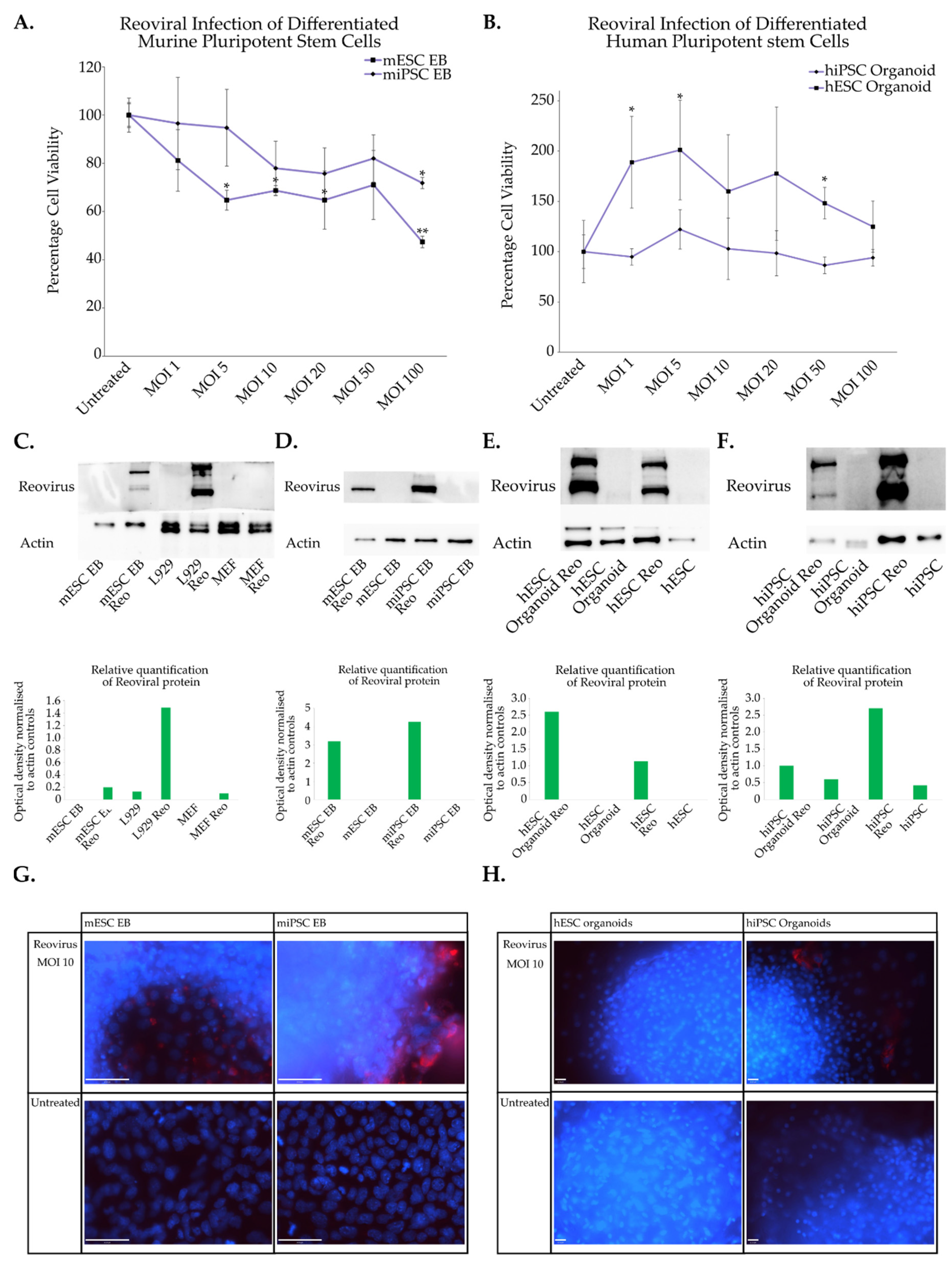
3.3. Reoviral Cytolysis in Differentiated Cells
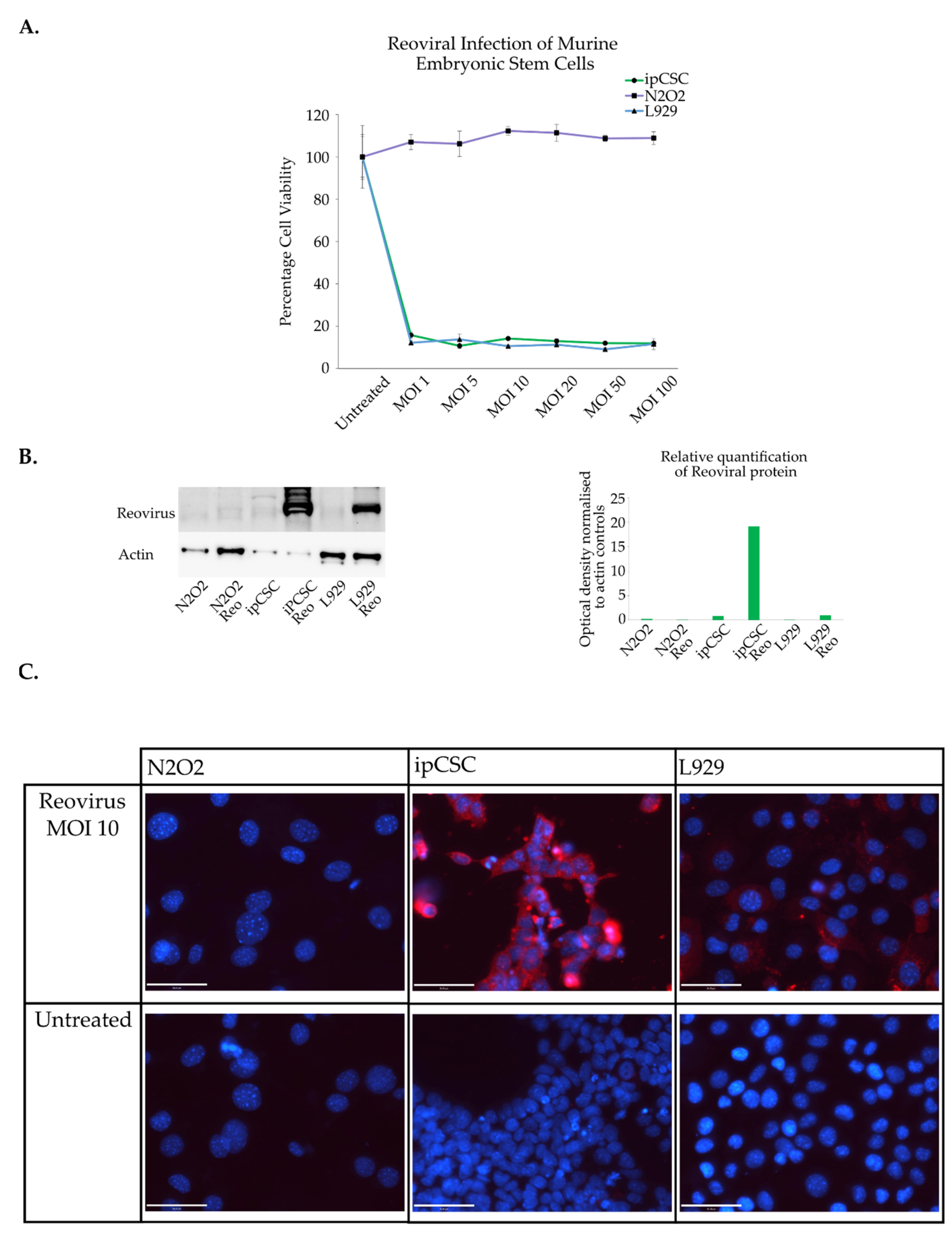
3.4. Induced Pluripotent Cancer Stem Cell Sensitivity to Reovirus
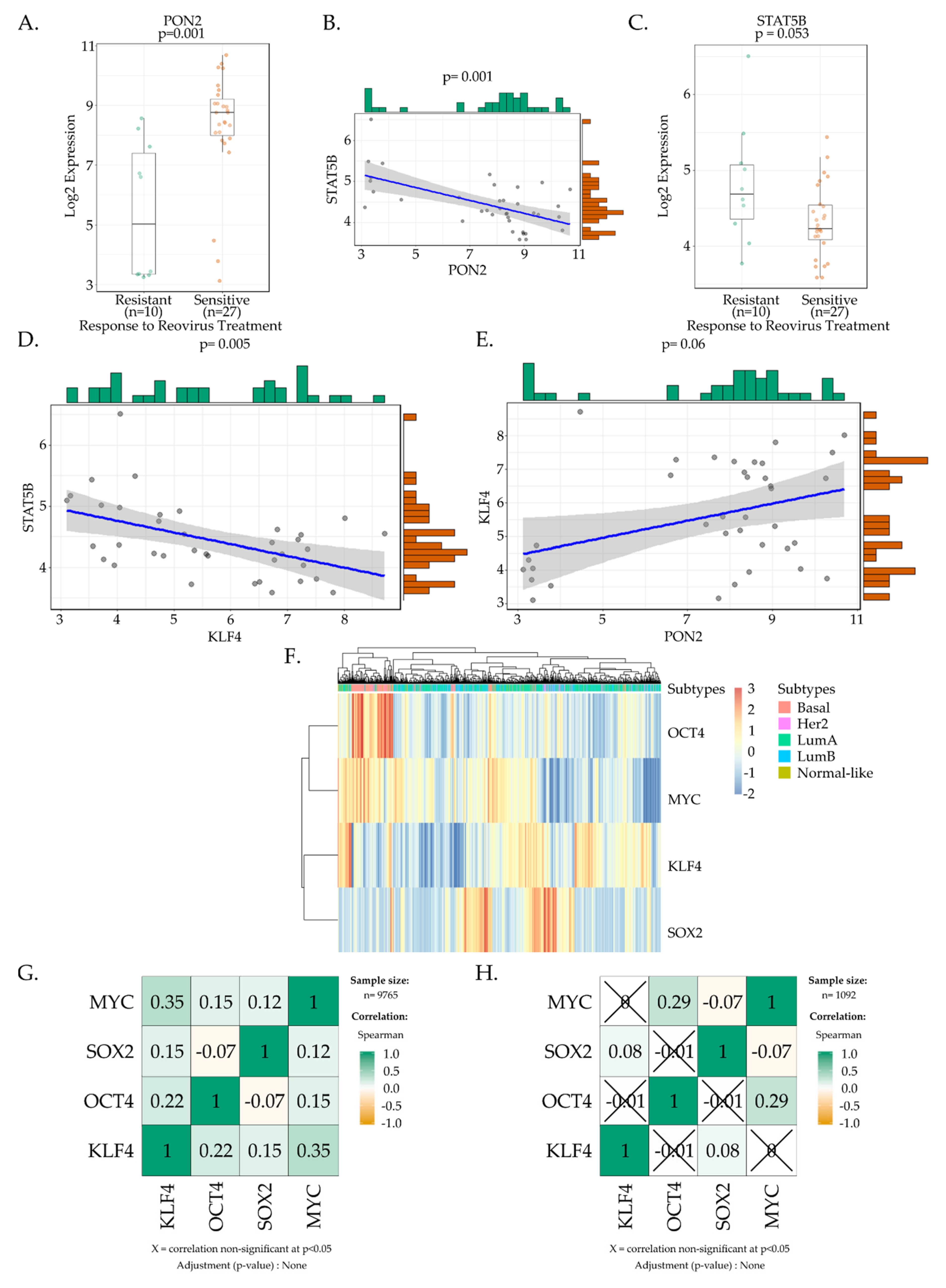
3.5. Bioinformatic Analysis of the Yamanaka Factors and Their Potential Role in Regulating Reoviral Sensitivity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Collichio, F.A.; Amatruda, T.; Senzer, N.N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Doleman, S.; Ye, Y.; et al. OPTiM: A randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J. Clin. Oncol. 2013, 31 (Suppl. S18), LBA9008. [Google Scholar] [CrossRef] [Green Version]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef] [Green Version]
- Oncolytics Biotech® Inc. Oncolytics Biotech® Inc. Announces Receipt of Orphan Drug Status from the EMA for Pancreatic Cancer. Available online: http://www.oncolyticsbiotech.com/news/press-release-details/2015/Oncolytics-Biotech-Inc-Announces-Receipt-of-Orphan-Drug-Status-from-the-EMA-for-Pancreatic-Cancer/default.aspx (accessed on 9 October 2015).
- Oncolytics Biotech® Inc. Oncolytics Biotech® Inc. Announces Receipt of Orphan Drug Designation from the U.S. FDA for Gastric Cancer. Available online: http://www.oncolyticsbiotech.com/news/press-release-details/2015/Oncolytics-Biotech-Inc-Announces-Receipt-of-Orphan-Drug-Designation-from-the-US-FDA-for-Gastric-Cancer/default.aspx (accessed on 9 October 2015).
- Galanis, E.; Markovic, S.N.; Suman, V.J.; Nuovo, G.J.; Vile, R.G.; Kottke, T.J.; Nevala, W.K.; Thompson, M.A.; Lewis, J.E.; Rumilla, K.M.; et al. Phase II trial of intravenous administration of Reolysin((R)) (Reovirus Serotype-3-dearing Strain) in patients with metastatic melanoma. Mol. Ther. 2012, 20, 1998–2003. [Google Scholar] [CrossRef] [Green Version]
- Coffey, M.C.; Strong, J.E.; Forsyth, P.A.; Lee, P.W. Reovirus therapy of tumors with activated Ras pathway. Science 1998, 282, 1332–1334. [Google Scholar] [CrossRef]
- Harrington, K.J.; Vile, R.G.; Melcher, A.; Chester, J.; Pandha, H.S. Clinical trials with oncolytic reovirus: Moving beyond phase I into combinations with standard therapeutics. Cytokine Growth Factor Rev. 2010, 21, 91–98. [Google Scholar] [CrossRef]
- Wilcox, M.E.; Yang, W.; Senger, D.; Rewcastle, N.B.; Morris, D.G.; Brasher, P.M.; Shi, Z.Q.; Johnston, R.N.; Nishikawa, S.; Lee, P.W.; et al. Reovirus as an oncolytic agent against experimental human malignant gliomas. J. Natl. Cancer Inst. 2001, 93, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.G.; Feng, X.; DiFrancesco, L.M.; Fonseca, K.; Forsyth, P.A.; Paterson, A.H.; Coffey, M.C.; Thompson, B. REO-001: A phase I trial of percutaneous intralesional administration of reovirus type 3 dearing (Reolysin(R)) in patients with advanced solid tumors. Investig. New Drugs 2013, 31, 696–706. [Google Scholar] [CrossRef]
- Forsyth, P.; Roldan, G.; George, D.; Wallace, C.; Palmer, C.A.; Morris, D.; Cairncross, G.; Matthews, M.V.; Markert, J.; Gillespie, Y.; et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol. Ther. 2008, 16, 627–632. [Google Scholar] [CrossRef]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields Virology; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Shatkin, A.J.; Sipe, J.D.; Loh, P. Separation of ten reovirus genome segments by polyacrylamide gel electrophoresis. J. Virol. 1968, 2, 986–991. [Google Scholar] [CrossRef] [Green Version]
- Hashiro, G.; Loh, P.C.; Yau, J.T. The preferential cytotoxicity of reovirus for certain transformed cell lines. Arch. Virol. 1977, 54, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.R.; Stanish, S.M.; Cox, D.C. Differential sensitivity of normal and transformed human cells to reovirus infectin. J. Virol. 1978, 28, 444–449. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Sachdev, E.; Mita, A.C.; Mita, M.M. Clinical development of reovirus for cancer therapy: An oncolytic virus with immune-mediated antitumor activity. World J. Methodol. 2016, 6, 25–42. [Google Scholar] [CrossRef]
- Clements, D.; Helson, E.; Gujar, S.A.; Lee, P.W. Reovirus in cancer therapy: An evidence-based review. Oncolytic Virotherapy 2014, 3, 69–82. [Google Scholar]
- Oncolytics Biotech® Inc. Oncolytics Biotech® Inc. Announces Receipt of Orphan Drug Status from the EMA for Ovarian Cancer. Available online: https://www.oncolyticsbiotech.com/press-releases/detail/340/oncolytics-biotech-inc-announces-receipt-of-orphan-drug (accessed on 30 July 2018).
- Oncolytics Biotech® Inc. Oncolytics Biotech® Receives Special Protocol Assessment Agreement from FDA for Phase 3 Clinical Trial of Pelareorep in Metastatic Breast Cancer. Available online: https://www.oncolyticsbiotech.com/press-releases/detail/412/oncolytics-biotech-receives-special-protocol-assessment (accessed on 9 October 2015).
- Oncolytics Biotech® Inc. Oncolytics Biotech® Inc. Announces FDA Fast Track Designation for REOLYSIN® in Metastatic Breast Cancer. Available online: https://www.oncolyticsbiotech.com/press-releases/detail/39/oncolytics-biotech-inc-announces-fda-fast-track (accessed on 9 October 2015).
- Joklik, W.K. Recent progress in reovirus research. Annu. Rev. Genet. 1985, 19, 537–575. [Google Scholar] [CrossRef] [PubMed]
- Masemann, D.; Boergeling, Y.; Ludwig, S. Employing RNA viruses to fight cancer: Novel insights into oncolytic virotherapy. Biol. Chem. 2017, 398, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chester, C.; Rajasekaran, N.; He, Z.; Kohrt, H.E. Strategic combinations: The future of oncolytic virotherapy with reovirus. Mol. Cancer Ther. 2016, 15, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Marchini, A.; Scott, E.; Rommelaere, J. Overcoming Barriers in Oncolytic Virotherapy with HDAC Inhibitors and Immune Checkpoint Blockade. Viruses 2016, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, R.; Miest, T.; Shashkova, E.V.; Barry, M.A. Reprogrammed viruses as cancer therapeutics: Targeted, armed and shielded. Nat. Rev. Microbiol. 2008, 6, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, S.; Hew, P.; Crosley, P.; Sharon, D.; Potts, K.; Agopsowicz, K.; Long, M.; Shi, C.; Hitt, M. Breast cancer gene therapy using an adenovirus encoding human IL-2 under control of mammaglobin promoter/enhancer sequences. Cancer Gene Ther. 2016, 23, 178–187. [Google Scholar] [CrossRef]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Miest, T.S.; Cattaneo, R. New viruses for cancer therapy: Meeting clinical needs. Nat. Rev. Microbiol. 2014, 12, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Antar, A.A.; Boehme, K.W.; Danthi, P.; Eby, E.A.; Guglielmi, K.M.; Holm, G.H.; Johnson, E.M.; Maginnis, M.S.; Naik, S. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 2007, 1, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Wollenberg, D.J.; Dautzenberg, I.J.; Ros, W.; Lipińska, A.D.; van den Hengel, S.K.; Hoeben, R.C. Replicating reoviruses with a transgene replacing the codons for the head domain of the viral spike. Gene Ther. 2015, 22, 267–279. [Google Scholar] [CrossRef]
- Bourhill, T.; Mori, Y.; Rancourt, D.E.; Shmulevitz, M.; Johnston, R.N. Going (reo) viral: Factors promoting successful reoviral oncolytic infection. Viruses 2018, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Strong, J.E.; Tang, D.; Lee, P.W. Evidence that the epidermal growth factor receptor on host cells confers reovirus infection efficiency. Virology 1993, 197, 405–411. [Google Scholar] [CrossRef]
- Strong, J.E.; Lee, P. The v-erbB oncogene confers enhanced cellular susceptibility to reovirus infection. J. Virol. 1996, 70, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Norman, K.L.; Lee, P.W. Reovirus as a novel oncolytic agent. J. Clin. Investig. 2000, 105, 1035–1038. [Google Scholar] [CrossRef] [Green Version]
- Strong, J.E.; Coffey, M.C.; Tang, D.; Sabinin, P.; Lee, P.W. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998, 17, 3351–3362. [Google Scholar] [CrossRef] [Green Version]
- Norman, K.L.; Hirasawa, K.; Yang, A.-D.; Shields, M.A.; Lee, P.W. Reovirus oncolysis: The Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 11099–11104. [Google Scholar] [CrossRef] [Green Version]
- Norman, K.L.; Farassati, F.; Lee, P.W.K. Oncolytic viruses and cancer therapy. Cytokine Growth Factor. Rev. 2001, 12, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Mita, M.M. Activated ras signaling pathways and reovirus oncolysis: An update on the mechanism of preferential reovirus replication in cancer cells. Front. Oncol. 2014, 4, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmulevitz, M.; Marcato, P.; Lee, P.W. Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer. Oncogene 2005, 24, 7720–7728. [Google Scholar] [CrossRef] [Green Version]
- Van Den Wollenberg, D.J.; Van Den Hengel, S.K.; Dautzenberg, I.J.; Kranenburg, O.; Hoeben, R.C. Modification of mammalian reoviruses for use as oncolytic agents. Expert. Opin. Biol. Ther. 2009, 9, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Thomis, D.C.; Samuel, C.E. Mechanism of interferon action: Evidence for intermolecular autophosphorylation and autoactivation of the interferon-induced, RNA-dependent protein kinase PKR. J. Virol. 1993, 67, 7695–7700. [Google Scholar] [CrossRef] [Green Version]
- Panniers, R.; Henshaw, E.C. A GDP/GTP exchange factor essential for eukaryotic initiation factor 2 cycling in Ehrlich ascites tumor cells and its regulation by eukaryotic initiation factor 2 phosphorylation. J. Biol. Chem. 1983, 258, 7928–7934. [Google Scholar] [CrossRef]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug. Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef]
- Krishnamoorthy, T.; Pavitt, G.D.; Zhang, F.; Dever, T.E.; Hinnebusch, A.G. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell Biol. 2001, 21, 5018–5030. [Google Scholar] [CrossRef] [Green Version]
- Terasawa, Y.; Hotani, T.; Katayama, Y.; Tachibana, M.; Mizuguchi, H.; Sakurai, F. Activity levels of cathepsins B and L in tumor cells are a biomarker for efficacy of reovirus-mediated tumor cell killing. Cancer Gene Ther. 2015, 22, 188–197. [Google Scholar] [CrossRef]
- Smakman, N.; van den Wollenberg, D.J.; Borel Rinkes, I.H.; Hoeben, R.C.; Kranenburg, O. Sensitization to apoptosis underlies KrasD12-dependent oncolysis of murine C26 colorectal carcinoma cells by reovirus T3D. J. Virol. 2005, 79, 14981–14985. [Google Scholar] [CrossRef] [Green Version]
- Smakman, N.; van den Wollenberg, D.J.; Elias, S.G.; Sasazuki, T.; Shirasawa, S.; Hoeben, R.C.; Borel Rinkes, I.H.; Kranenburg, O. KRAS(D13) Promotes apoptosis of human colorectal tumor cells by ReovirusT3D and oxaliplatin but not by tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. 2006, 66, 5403–5408. [Google Scholar] [CrossRef] [Green Version]
- Van Houdt, W.J.; Smakman, N.; van den Wollenberg, D.J.; Emmink, B.L.; Veenendaal, L.M.; van Diest, P.J.; Hoeben, R.C.; Borel Rinkes, I.H.; Kranenburg, O. Transient infection of freshly isolated human colorectal tumor cells by reovirus T3D intermediate subviral particles. Cancer Gene Ther. 2008, 15, 284–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Samuel, C.E. Protein kinase PKR plays a stimulus- and virus-dependent role in apoptotic death and virus multiplication in human cells. J. Virol. 2007, 81, 8192–8200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twigger, K.; Roulstone, V.; Kyula, J.; Karapanagiotou, E.M.; Syrigos, K.N.; Morgan, R.; White, C.; Bhide, S.; Nuovo, G.; Coffey, M.; et al. Reovirus exerts potent oncolytic effects in head and neck cancer cell lines that are independent of signalling in the EGFR pathway. BMC Cancer 2012, 12, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.A.; Schmechel, S.C.; Williams, B.R.; Silverman, R.H.; Schiff, L.A. Involvement of the interferon-regulated antiviral proteins PKR and RNase L in reovirus-induced shutoff of cellular translation. J. Virol. 2005, 79, 2240–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.A.; Schmechel, S.C.; Raghavan, A.; Abelson, M.; Reilly, C.; Katze, M.G.; Kaufman, R.J.; Bohjanen, P.R.; Schiff, L.A. Reovirus induces and benefits from an integrated cellular stress response. J. Virol. 2006, 80, 2019–2033. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Vähä-Koskela, M.J.; Heikkilä, J.E.; Hinkkanen, A.E. Oncolytic viruses in cancer therapy. Cancer Lett. 2007, 254, 178–216. [Google Scholar] [CrossRef]
- Bierman, H.R.; Crile, D.M.; Dod, K.S.; Kelly, K.H.; Petrakis, N.L.; White, L.P.; Shimkin, M.B. Remissions in leukemia of childhood following acute infectious disease: Staphylococcus and streptococcus, varicella, and feline panleukopenia. Cancer 1953, 6, 591–605. [Google Scholar] [CrossRef]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W. Oncolytic reovirus effectively targets breast cancer stem cells. Mol. Ther. 2009, 17, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Cripe, T.P.; Wang, P.Y.; Marcato, P.; Mahller, Y.Y.; Lee, P.W. Targeting cancer-initiating cells with oncolytic viruses. Mol. Ther. 2009, 17, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Garant, K.A.; zur Nieden, N.I.; Alain, T.; Loken, S.D.; Urbanski, S.J.; Forsyth, P.A.; Rancourt, D.E.; Lee, P.W.; Johnston, R.N. Attenuated reovirus displays oncolysis with reduced host toxicity. Br. J. Cancer 2011, 104, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, T.G.; Kockeritz, L.; Szrajber, M.R.; Muller, W.J.; Hassell, J.A. The pea3 subfamily ets genes are required for HER2/Neu-mediated mammary oncogenesis. Curr. Biol. 2001, 11, 1739–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafa, M.; Sjonnesen, K.; Yamashita, A.; Liu, S.; Michalak, M.; Kallos, M.S.; Rancourt, D.E. Expansion and long-term maintenance of induced pluripotent stem cells in stirred suspension bioreactors. J. Tissue Eng. Regen. Med. 2012, 6, 462–472. [Google Scholar] [CrossRef]
- Conley, B.J.; Denham, M.; Gulluyan, L.; Olsson, F.; Cole, T.J.; Mollard, R. Mouse Embryonic Stem Cell Derivation, and Mouse and Human Embryonic Stem Cell Culture and Differentiation as Embryoid Bodies. Curr. Protoc. Cell Biol. 2005, 28, 23.2.1–23.2.22. [Google Scholar] [CrossRef]
- Meng, G.; Liu, S.; Rancourt, D.E. Rapid isolation of undifferentiated human pluripotent stem cells from extremely differentiated colonies. Stem Cells Dev. 2011, 20, 583–591. [Google Scholar] [CrossRef]
- Kurosawa, H. Methods for inducing embryoid body formation: In vitro differentiation system of embryonic stem cells. J. Biosci. Bioeng. 2007, 103, 389–398. [Google Scholar] [CrossRef]
- Keller, G.M. In vitro differentiation of embryonic stem cells. Curr. Opin. Cell Biol. 1995, 7, 862–869. [Google Scholar] [CrossRef]
- Heidariyan, Z.; Ghanian, M.H.; Ashjari, M.; Farzaneh, Z.; Najarasl, M.; Rezaei Larijani, M.; Piryaei, A.; Vosough, M.; Baharvand, H. Efficient and cost-effective generation of hepatocyte-like cells through microparticle-mediated delivery of growth factors in a 3D culture of human pluripotent stem cells. Biomaterials 2018, 159, 174–188. [Google Scholar] [CrossRef]
- Vega-Avila, E.; Pugsley, M.K. An overview of colorimetric assay methods used to assess survival or proliferation of mammalian cells. Proc. West. Pharmacol. Soc. 2011, 54, 10–14. [Google Scholar] [PubMed]
- Kueng, W.; Silber, E.; Eppenberger, U. Quantification of cells cultured on 96-well plates. Anal. Biochem. 1989, 182, 16–19. [Google Scholar] [CrossRef]
- Alain, T.; Kim, M.; Johnston, R.; Urbanski, S.; Kossakowska, A.; Forsyth, P.; Lee, P. The oncolytic effect in vivo of reovirus on tumour cells that have survived reovirus cell killing in vitro. Br. J. Cancer 2006, 95, 1020–1027. [Google Scholar] [CrossRef] [Green Version]
- Lichtman, J.W.; Conchello, J.-A. Fluorescence microscopy. Nat. Methods 2005, 2, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Network, C.G.A.R. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Banijamali, R.S.; Soleimanjahi, H.; Soudi, S.; Karimi, H.; Abdoli, A.; Khorrami, S.M.S.; Zandi, K. Kinetics of Oncolytic Reovirus T3D Replication and Growth Pattern in Mesenchymal Stem Cells. Cell J. 2020, 22, 283–292. [Google Scholar]
- Park, J.-S.; Kim, M. Reovirus safety study for proliferation and differentiation of human adipose-derived mesenchymal stem cells. J. Microbiol. 2017, 55, 75–79. [Google Scholar] [CrossRef]
- Hotta, A.; Cheung, A.Y.; Farra, N.; Vijayaragavan, K.; Séguin, C.A.; Draper, J.S.; Pasceri, P.; Maksakova, I.A.; Mager, D.L.; Rossant, J. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat. Methods 2009, 6, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Schoenhals, M.; Kassambara, A.; De Vos, J.; Hose, D.; Moreaux, J.; Klein, B. Embryonic stem cell markers expression in cancers. Biochem. Biophys. Res. Commun. 2009, 383, 157–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, C.T.; Webster, M.A.; Schaller, M.; Parsons, T.J.; Cardiff, R.D.; Muller, W.J. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10578–10582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiecker, L.; Kindler, T.; Horke, S.; Witte, I. Paraoxonase-2 alters hematopoietic stem cell differentiation through redox signalling. Exp. Hematol. 2017, 53, S65. [Google Scholar] [CrossRef]
- Yuan, J.; Devarajan, A.; Moya-Castro, R.; Zhang, M.; Evans, S.; Bourquard, N.; Dias, P.; Lacout, C.; Vainchenker, W.; Reddy, S.T. Putative innate immunity of antiatherogenic paraoxanase-2 via STAT5 signal transduction in HIV-1 infection of hematopoietic TF-1 cells and in SCID-hu mice. J. Stem Cells 2010, 5, 43–48. [Google Scholar]
- Thirukkumaran, C.; Morris, D.G. Oncolytic viral therapy using reovirus. In Gene Therapy of Solid Cancers; Springer: Berlin/Heidelberg, Germany, 2015; pp. 187–223. [Google Scholar]
- Choi, A.H.; O’Leary, M.P.; Fong, Y.; Chen, N.G. From benchtop to bedside: A review of oncolytic virotherapy. Biomedicines 2016, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Kemp, V.; Hoeben, R.C.; Van den Wollenberg, D.J. Exploring reovirus plasticity for improving its use as oncolytic virus. Viruses 2016, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, A.; Johnston, R.N.; Shmulevitz, M. Potential for improving potency and specificity of reovirus oncolysis with next-generation reovirus variants. Viruses 2015, 7, 6251–6278. [Google Scholar] [CrossRef] [Green Version]
- Thirukkumaran, C.M.; Luider, J.M.; Stewart, D.A.; Cheng, T.; Lupichuk, S.M.; Nodwell, M.J.; Russell, J.A.; Auer, I.A.; Morris, D.G. Reovirus oncolysis as a novel purging strategy for autologous stem cell transplantation. Blood 2003, 102, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Collins, D.C.; Sundar, R.; Lim, J.S.; Yap, T.A. Towards precision medicine in the clinic: From biomarker discovery to novel therapeutics. Trends Pharmacol. Sci. 2017, 38, 25–40. [Google Scholar] [CrossRef]
- Miao, D.; Van Allen, E.M. Genomic determinants of cancer immunotherapy. Curr. Opin. Immunol. 2016, 41, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef]
- Alain, T.; Kim, T.S.; Lun, X.; Liacini, A.; Schiff, L.A.; Senger, D.L.; Forsyth, P.A. Proteolytic disassembly is a critical determinant for reovirus oncolysis. Mol. Ther. 2007, 15, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Barton, E.S.; Forrest, J.C.; Connolly, J.L.; Chappell, J.D.; Liu, Y.; Schnell, F.J.; Nusrat, A.; Parkos, C.A.; Dermody, T.S. Junction adhesion molecule is a receptor for reovirus. Cell 2001, 104, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maginnis, M.S.; Forrest, J.C.; Kopecky-Bromberg, S.A.; Dickeson, S.K.; Santoro, S.A.; Zutter, M.M.; Nemerow, G.R.; Bergelson, J.M.; Dermody, T.S. Beta1 integrin mediates internalization of mammalian reovirus. J. Virol. 2006, 80, 2760–2770. [Google Scholar] [CrossRef] [Green Version]
- Theunissen, T.W.; Jaenisch, R. Molecular control of induced pluripotency. Cell Stem Cell 2014, 14, 720–734. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Nishikawa, S.G.; Fratiloiu, A.R.; Tsutsui, M.; Kataoka, H.; Joh, T.; Johnston, R.N. Modulation of Reoviral Cytolysis (I): Combination Therapeutics. Viruses 2023, 15, 1472. [Google Scholar] [CrossRef]
- Bortoletto, A.S.; Parchem, R.J. KRAS Hijacks the miRNA Regulatory Pathway in Cancer. Cancer Res. 2023, 83, 1563–1572. [Google Scholar] [CrossRef]
- Inoue, C.; Negoro, R.; Takayama, K.; Mizuguchi, H.; Sakurai, F. Asymmetric profiles of infection and innate immunological responses in human iPS cell-derived small intestinal epithelial-like cell monolayers following infection with mammalian reovirus. Virus Res. 2021, 296, 198–334. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourhill, T.; Rohani, L.; Kumar, M.; Bose, P.; Rancourt, D.; Johnston, R.N. Modulation of Reoviral Cytolysis (II): Cellular Stemness. Viruses 2023, 15, 1473. https://doi.org/10.3390/v15071473
Bourhill T, Rohani L, Kumar M, Bose P, Rancourt D, Johnston RN. Modulation of Reoviral Cytolysis (II): Cellular Stemness. Viruses. 2023; 15(7):1473. https://doi.org/10.3390/v15071473
Chicago/Turabian StyleBourhill, Tarryn, Leili Rohani, Mehul Kumar, Pinaki Bose, Derrick Rancourt, and Randal N. Johnston. 2023. "Modulation of Reoviral Cytolysis (II): Cellular Stemness" Viruses 15, no. 7: 1473. https://doi.org/10.3390/v15071473