
Tumour Necrosis Factor-α, Chemokines, and Leukocyte Infiltrate Are Biomarkers for Pathology in the Brains of Venezuelan Equine Encephalitis (VEEV)-Infected Mice
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Virus
2.2. In Vivo Studies
2.3. Plaque Assay
2.4. Immunological Methods
2.5. Histopathology Methods
2.6. Statistical Analysis
3. Results
3.1. The Progression of VEEV Disease in Infected Mice
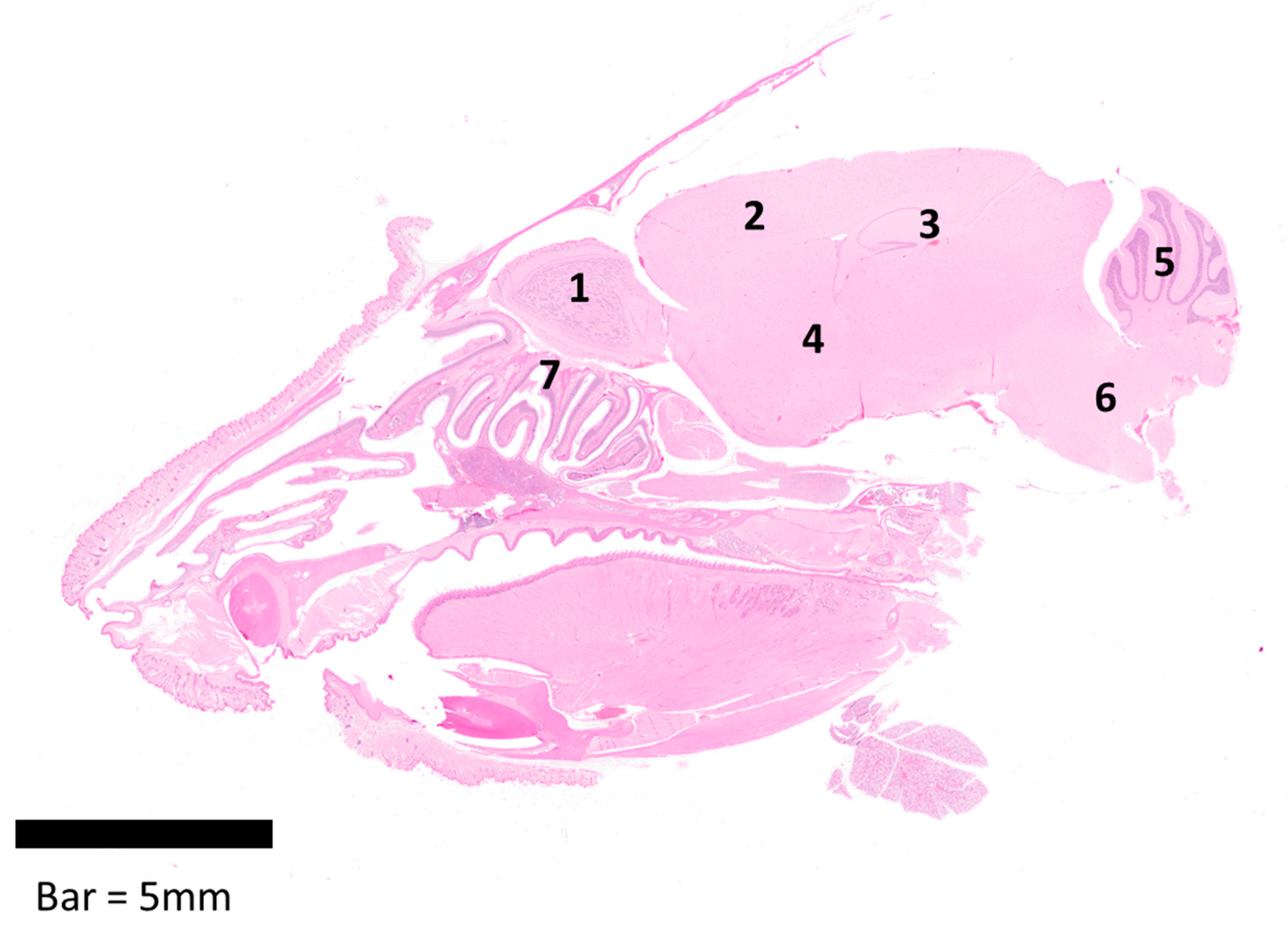
3.2. Pathology and Virus Distribution in Brain/Encephalon and Nasal Cavity
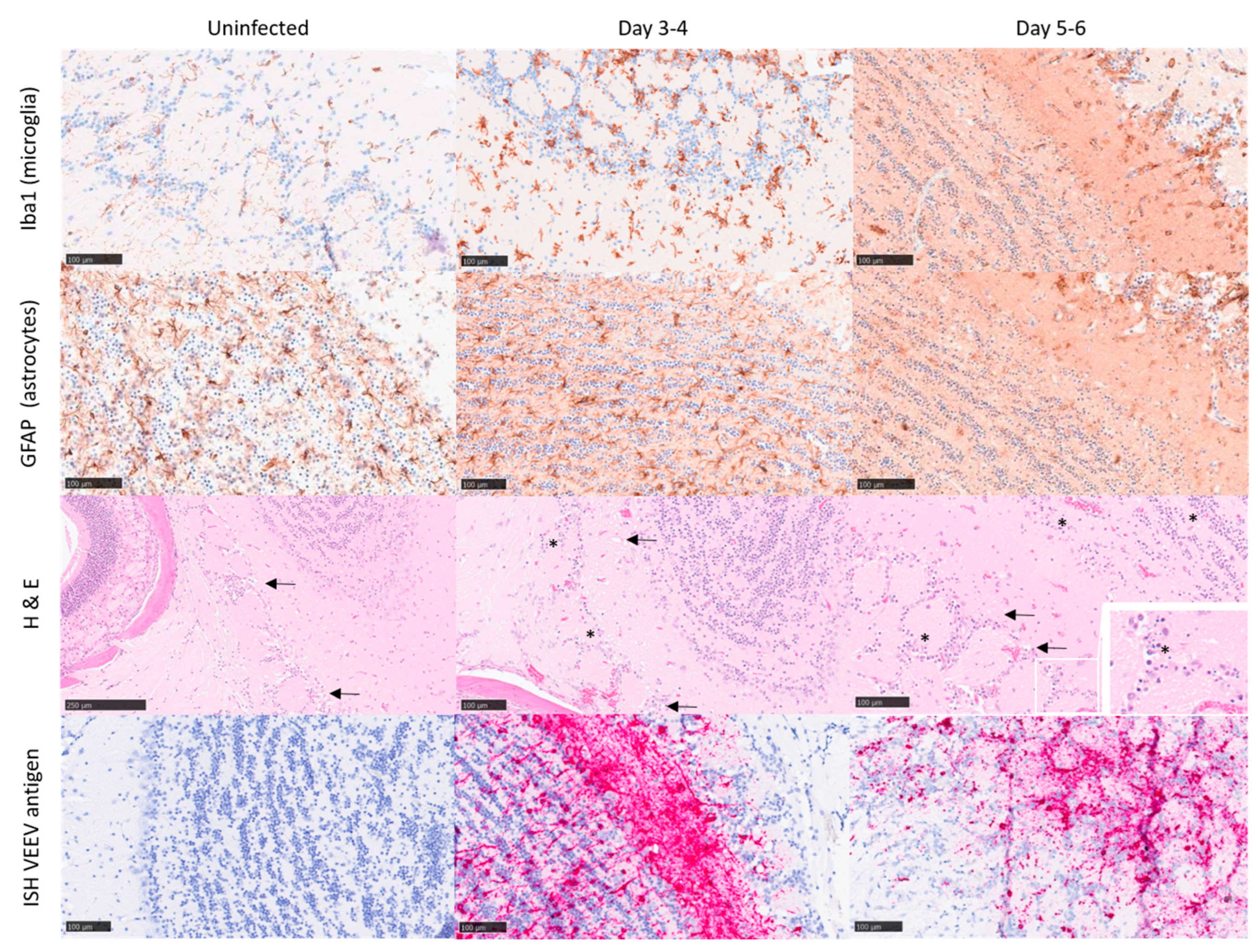
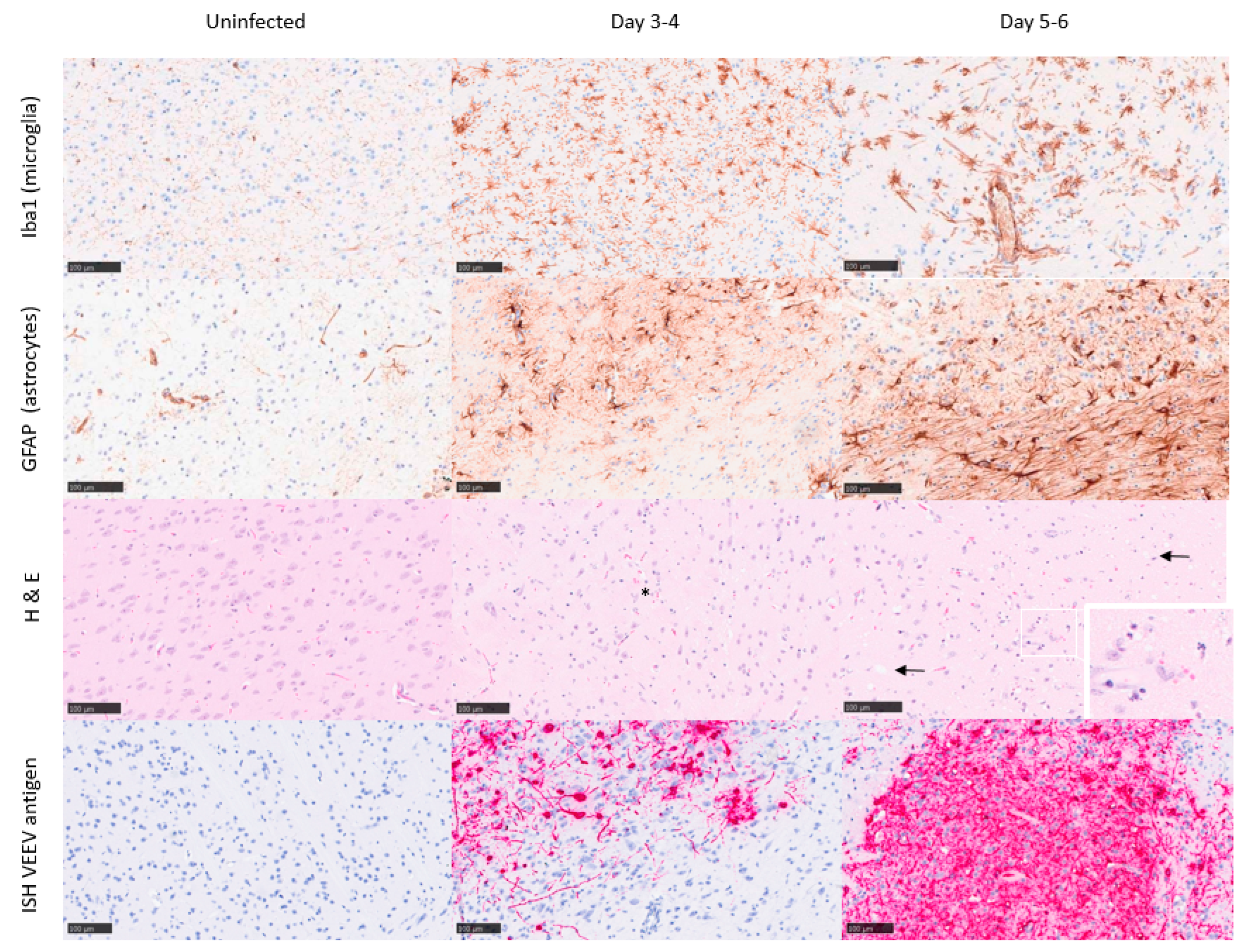
3.3. Inflammatory Response to Infection
3.4. Certain Inflammatory Markers Are Biomarkers for Neuropathology
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daza, E.; Frias, V.; Alcola, A.; Lopez, I.; Bruzon, I.; Montero, J.T.; Alvarez, G.; Garcia, M.A.; Rodriguez, R.; Boschell, J.; et al. Venezuelan equine encephalitis—Colombia, 1995. MMWR Morb. Mortal. Wkly. Rep. 1995, 44, 721–724. [Google Scholar]
- Hanson, R.P.; Sulkin, S.E.; Beuscher, E.L.; Hammon, W.M.; McKinney, R.W.; Work, T.H. Arbovirus infections of laboratory workers. Extent of problem emphasizes the need for more effective measures to reduce hazards. Science 1967, 158, 1283–1286. [Google Scholar] [CrossRef] [PubMed]
- Honnold, S.P.; Mossel, E.C.; Dupuy, L.C.; Morazzani, E.M.; Martin, S.S.; Hart, M.K.; Ludwig, G.V.; Parker, M.D.; Smith, J.F.; Reed, D.S. Alphavirus Encephalitides. In Medical Aspects of Biological Warfare; Bozue, J., Cote, C.K., Glass, P.J., Eds.; Borden Institute: San Antonio, TX, USA, 2018; p. 483. [Google Scholar]
- Special Immunizations Program. Available online: https://www.usammda.army.mil/index.cfm/fhp/immunizations_program (accessed on 20 April 2023).
- Pittman, P.R.; Brown, E.S.; Chambers, M.S. Medical Countermeasures. In Medical Aspects of Biological Warfare; Bozue, J., Cote, C.K., Glass, P.J., Eds.; Borden Institute: San Antonio, TX, USA, 2018; pp. 773–776. [Google Scholar]
- Stromberg, Z.R.; Fischer, W.; Bradfute, S.B.; Kubicek-Sutherland, J.Z.; Hraber, P. Vaccine Advances against Venezuelan, Eastern, and Western Equine Encephalitis Viruses. Vaccines 2020, 8, 273. [Google Scholar] [CrossRef]
- Burke, C.W.; Erwin-Cohen, R.A.; Goodson, A.I.; Wilhelmsen, C.; Edmundson, J.A.; White, C.E.; Glass, P.J. Efficacy of Western, Eastern, and Venezuelan Equine Encephalitis (WEVEE) Virus-Replicon Particle (VRP) Vaccine against WEEV in a Non-Human Primate Animal Model. Viruses 2022, 14, 1502. [Google Scholar] [CrossRef]
- Tretyakova, I.; Plante, K.S.; Rossi, S.L.; Lawrence, W.S.; Peel, J.E.; Gudjohnsen, S.; Wang, E.; Mirchandani, D.; Tibbens, A.; Lamichhane, T.N.; et al. Venezuelan equine encephalitis vaccine with rearranged genome resists reversion and protects non-human primates from viremia after aerosol challenge. Vaccine 2020, 38, 3378–3386. [Google Scholar] [CrossRef] [PubMed]
- Reed, D.S.; Glass, P.J.; Bakken, R.R.; Barth, J.F.; Lind, C.M.; da Silva, L.; Hart, M.K.; Rayner, J.; Alterson, K.; Custer, M.; et al. Combined alphavirus replicon particle vaccine induces durable and cross-protective immune responses against equine encephalitis viruses. J. Virol. 2014, 88, 12077–12086. [Google Scholar] [CrossRef] [Green Version]
- Coates, E.E.; Edupuganti, S.; Chen, G.L.; Happe, M.; Strom, L.; Widge, A.; Florez, M.B.; Cox, J.H.; Gordon, I.; Plummer, S.; et al. Safety and immunogenicity of a trivalent virus-like particle vaccine against western, eastern, and Venezuelan equine encephalitis viruses: A phase 1, open-label, dose-escalation, randomised clinical trial. Lancet. Infect. Dis. 2022, 22, 1210–1220. [Google Scholar] [CrossRef]
- Hannaman, D.; Dupuy, L.C.; Ellefsen, B.; Schmaljohn, C.S. A Phase 1 clinical trial of a DNA vaccine for Venezuelan equine encephalitis delivered by intramuscular or intradermal electroporation. Vaccine 2016, 34, 3607–3612. [Google Scholar] [CrossRef]
- Phillpotts, R.J.; Jones, L.D.; Howard, S.C. Monoclonal antibody protects mice against infection and disease when given either before or up to 24 h after airborne challenge with virulent Venezuelan equine encephalitis virus. Vaccine 2002, 20, 1497–1504. [Google Scholar] [CrossRef]
- Phillpotts, R.J.; Jones, L.D.; Lukaszewski, R.A.; Lawrie, C.; Brooks, T.J. Antibody and interleukin-12 treatment in murine models of encephalitogenic flavivirus (St. Louis encephalitis, tick-borne encephalitis) and alphavirus (Venezuelan equine encephalitis) infection. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2003, 23, 47–50. [Google Scholar] [CrossRef]
- Hunt, A.R.; Frederickson, S.; Hinkel, C.; Bowdish, K.S.; Roehrig, J.T. A humanized murine monoclonal antibody protects mice either before or after challenge with virulent Venezuelan equine encephalomyelitis virus. J. Gen. Virol. 2006, 87 Pt 9, 2467–2476. [Google Scholar] [CrossRef] [PubMed]
- Phillpotts, R.J. Venezuelan equine encephalitis virus complex-specific monoclonal antibody provides broad protection, in murine models, against airborne challenge with viruses from serogroups I, II and III. Virus Res. 2006, 120, 107–112. [Google Scholar] [CrossRef]
- O’Brien, L.M.; Underwood-Fowler, C.D.; Goodchild, S.A.; Phelps, A.L.; Phillpotts, R.J. Development of a novel monoclonal antibody with reactivity to a wide range of Venezuelan equine encephalitis virus strains. Virol. J. 2009, 6, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.G.; Phelps, A.L.; Jager, S.; Chau, D.; Hu, C.C.; O’Brien, L.M.; Perkins, S.D.; Gates, A.J.; Phillpotts, R.J.; Nagata, L.P. A recombinant humanized monoclonal antibody completely protects mice against lethal challenge with Venezuelan equine encephalitis virus. Vaccine 2010, 28, 5558–5564. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.D.; Buckley, M.J.; Melanson, V.R.; Glass, P.J.; Norwood, D.; Hart, M.K. Antibody to the E3 glycoprotein protects mice against lethal venezuelan equine encephalitis virus infection. J. Virol. 2010, 84, 12683–12690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodchild, S.A.; O’Brien, L.M.; Steven, J.; Muller, M.R.; Lanning, O.J.; Logue, C.H.; D’Elia, R.V.; Phillpotts, R.J.; Perkins, S.D. A humanised murine monoclonal antibody with broad serogroup specificity protects mice from challenge with Venezuelan equine encephalitis virus. Antivir. Res. 2011, 90, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.R.; Bowen, R.A.; Frederickson, S.; Maruyama, T.; Roehrig, J.T.; Blair, C.D. Treatment of mice with human monoclonal antibody 24h after lethal aerosol challenge with virulent Venezuelan equine encephalitis virus prevents disease but not infection. Virology 2011, 414, 146–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, L.M.; Goodchild, S.A.; Phillpotts, R.J.; Perkins, S.D. A humanised murine monoclonal antibody protects mice from Venezuelan equine encephalitis virus, Everglades virus and Mucambo virus when administered up to 48 h after airborne challenge. Virology 2012, 426, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Burke, C.W.; Froude, J.W.; Rossi, F.; White, C.E.; Moyer, C.L.; Ennis, J.; Pitt, M.L.; Streatfield, S.; Jones, R.M.; Musiychuk, K.; et al. Therapeutic monoclonal antibody treatment protects nonhuman primates from severe Venezuelan equine encephalitis virus disease after aerosol exposure. PLoS Pathog. 2019, 15, e1008157. [Google Scholar] [CrossRef] [Green Version]
- Kafai, N.M.; Williamson, L.E.; Binshtein, E.; Sukupolvi-Petty, S.; Gardner, C.L.; Liu, J.; Mackin, S.; Kim, A.S.; Kose, N.; Carnahan, R.H.; et al. Neutralizing antibodies protect mice against Venezuelan equine encephalitis virus aerosol challenge. J. Exp. Med. 2022, 219, e20212532. [Google Scholar] [CrossRef]
- Lukaszewski, R.A.; Brooks, T.J. Pegylated alpha interferon is an effective treatment for virulent venezuelan equine encephalitis virus and has profound effects on the host immune response to infection. J. Virol. 2000, 74, 5006–5015. [Google Scholar] [CrossRef] [PubMed]
- Risner, K.; Ahmed, A.; Bakovic, A.; Kortchak, S.; Bhalla, N.; Narayanan, A. Efficacy of FDA-Approved Anti-Inflammatory Drugs Against Venezuelan Equine Encephalitis Virus Infection. Viruses 2019, 11, 1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RECOVERY Randomised Evaluation of COVID-19 Therapy. Available online: https://www.recoverytrial.net/ (accessed on 20 April 2023).
- Montiel, M.; Bonilla, E.; Valero, N.; Mosquera, J.; Espina, L.M.; Quiroz, Y.; Alvarez-Mon, M. Melatonin decreases brain apoptosis, oxidative stress, and CD200 expression and increased survival rate in mice infected by Venezuelan equine encephalitis virus. Antivir. Chem. Chemother. 2016, 24, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Blakely, P.K.; Huber, A.K.; Irani, D.N. Type-1 angiotensin receptor signaling in central nervous system myeloid cells is pathogenic during fatal alphavirus encephalitis in mice. J. Neuroinflamm. 2016, 13, 196. [Google Scholar] [CrossRef] [Green Version]
- Irani, D.N.; Prow, N.A. Neuroprotective interventions targeting detrimental host immune responses protect mice from fatal alphavirus encephalitis. J. Neuropathol. Exp. Neurol. 2007, 66, 533–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, A.; Brooke, C.B.; Whitmore, A.C.; Johnston, R.E. The role of the blood-brain barrier during Venezuelan equine encephalitis virus infection. J. Virol. 2011, 85, 10682–10690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Bhattacharya, B.; Puri, R.K.; Maheshwari, R.K. Venezuelan equine encephalitis virus infection causes modulation of inflammatory and immune response genes in mouse brain. BMC Genom. 2008, 9, 289. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Bhomia, M.; Honnold, S.P.; Maheshwari, R.K. Role of adhesion molecules and inflammation in Venezuelan equine encephalitis virus infected mouse brain. Virol. J. 2011, 8, 197. [Google Scholar] [CrossRef] [Green Version]
- Schoneboom, B.A.; Catlin, K.M.K.; Marty, A.M.; Grieder, F.B. Inflammation is a component of neurodegeneration in response to Venezuelan equine encephalitis virus infection in mice. J. Neuroimmunol. 2000, 109, 132–146. [Google Scholar] [CrossRef]
- Hollidge, B.S.; Cohen, C.A.; Akuoku Frimpong, J.; Badger, C.V.; Dye, J.M.; Schmaljohn, C.S. Toll-like receptor 4 mediates blood-brain barrier permeability and disease in C3H mice during Venezuelan equine encephalitis virus infection. Virulence 2021, 12, 430–443. [Google Scholar] [CrossRef]
- Bocan, T.M.; Stafford, R.G.; Brown, J.L.; Akuoku Frimpong, J.; Basuli, F.; Hollidge, B.S.; Zhang, X.; Raju, N.; Swenson, R.E.; Smith, D.R. Characterization of Brain Inflammation, Apoptosis, Hypoxia, Blood-Brain Barrier Integrity and Metabolism in Venezuelan Equine Encephalitis Virus (VEEV TC-83) Exposed Mice by In Vivo Positron Emission Tomography Imaging. Viruses 2019, 11, 1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, P.C.; Trgovcich, J.; Davis, N.L.; Johnston, R.E. Immunopathogenesis and immune modulation of Venezuelan equine encephalitis virus-induced disease in the mouse. Virology 2001, 284, 190–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, K.E.; Twenhafel, N.A. REVIEW PAPER: Pathology of animal models of alphavirus encephalitis. Vet. Pathol. 2010, 47, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS immune privilege: Hiding in plain sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef]
- Phillpotts, R.J.; O’Brien, L.; Appleton, R.E.; Carr, S.; Bennett, A. Intranasal immunisation with defective adenovirus serotype 5 expressing the Venezuelan equine encephalitis virus E2 glycoprotein protects against airborne challenge with virulent virus. Vaccine 2005, 23, 1615–1623. [Google Scholar] [CrossRef]
- Cain, M.D.; Salimi, H.; Gong, Y.; Yang, L.; Hamilton, S.L.; Heffernan, J.R.; Hou, J.; Miller, M.J.; Klein, R.S. Virus entry and replication in the brain precedes blood-brain barrier disruption during intranasal alphavirus infection. J. Neuroimmunol. 2017, 308, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Weaver, S.C.; Ferro, C.; Barrera, R.; Boshell, J.; Navarro, J.C. Venezuelan equine encephalitis. Annu. Rev. Entomol. 2004, 49, 141–174. [Google Scholar] [CrossRef]
- Gadani, S.P.; Cronk, J.C.; Norris, G.T.; Kipnis, J. IL-4 in the Brain: A Cytokine to Remember. J. Immunol. 2012, 189, 4213. [Google Scholar] [CrossRef] [Green Version]
- Miossec, P.; Korn, T.; Kuchroo, V.K. Interleukin-17 and type 17 helper T cells. N. Engl. J. Med. 2009, 361, 888–898. [Google Scholar] [CrossRef] [Green Version]
- Daniels, B.P.; Holman, D.W.; Cruz-Orengo, L.; Jujjavarapu, H.; Durrant, D.M.; Klein, R.S. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. mBio 2014, 5, e01476-14. [Google Scholar] [CrossRef] [Green Version]
- Durrant, D.M.; Ghosh, S.; Klein, R.S. The Olfactory Bulb: An Immunosensory Effector Organ during Neurotropic Viral Infections. ACS Chem. Neurosci. 2016, 7, 464–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laws, T.R.; Taylor, A.W.; Russell, P.; Williamson, D. The treatment of melioidosis: Is there a role for repurposed drugs? A proposal and review. Expert Rev. Anti-Infect. Ther. 2019, 17, 957–967. [Google Scholar] [CrossRef] [PubMed]
- RECOVERY Collaborative Group. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2020, 384, 693–704. [Google Scholar]
- Newton, R.; Holden, N.S. Separating transrepression and transactivation: A distressing divorce for the glucocorticoid receptor? Mol. Pharmacol. 2007, 72, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Consortia, R. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2021, 397, 1637–1645. [Google Scholar]
- Jones, S.A.; Takeuchi, T.; Aletaha, D.; Smolen, J.; Choy, E.H.; McInnes, I. Interleukin 6: The biology behind the therapy. Consid. Med. 2018, 2, 2. [Google Scholar] [CrossRef]
- Steinskog, E.S.; Sagstad, S.J.; Wagner, M.; Karlsen, T.V.; Yang, N.; Markhus, C.E.; Yndestad, S.; Wiig, H.; Eikesdal, H.P. Impaired lymphatic function accelerates cancer growth. Oncotarget 2016, 7, 45789–45802. [Google Scholar] [CrossRef] [Green Version]
- Fei, Y.; Wang, W.; Kwiecinski, J.; Josefsson, E.; Pullerits, R.; Jonsson, I.M.; Magnusson, M.; Jin, T. The combination of a tumor necrosis factor inhibitor and antibiotic alleviates staphylococcal arthritis and sepsis in mice. J. Infect. Dis. 2011, 204, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Dhimolea, E. Canakinumab. MAbs 2010, 2, 3–13. [Google Scholar] [CrossRef]
- Arend, W.P. Interleukin-1 receptor antagonist: Discovery, structure and properties. Prog. Growth Factor Res. 1990, 2, 193–205. [Google Scholar] [CrossRef]
- Ceballos-Olvera, I.; Sahoo, M.; Miller, M.A.; Del Barrio, L.; Re, F. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1beta is deleterious. PLoS Pathog. 2011, 7, e1002452. [Google Scholar] [CrossRef] [PubMed]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran. J. Pharm. Res. IJPR 2011, 10, 655–683. [Google Scholar] [PubMed]
- Consortia, R. Aspirin in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2022, 399, 143–151. [Google Scholar]
- Bedoui, Y.; Septembre-Malaterre, A.; Giry, C.; Jaffar-Bandjee, M.C.; Selambarom, J.; Guiraud, P.; Gasque, P. Robust COX-2-mediated prostaglandin response may drive arthralgia and bone destruction in patients with chronic inflammation post-chikungunya. PLoS Negl. Trop. Dis. 2021, 15, e0009115. [Google Scholar] [CrossRef] [PubMed]
- Lehman, C.W.; Smith, A.; Kelly, J.; Jacobs, J.L.; Dinman, J.D.; Kehn-Hall, K. EGR1 Upregulation during Encephalitic Viral Infections Contributes to Inflammation and Cell Death. Viruses 2022, 14, 1210. [Google Scholar] [CrossRef]
- Consortia, R. Baricitinib in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial and updated meta-analysis. Lancet 2022, 400, 359–368. [Google Scholar]
- van Vollenhoven, R.F. Small molecular compounds in development for rheumatoid arthritis. Curr. Opin. Rheumatol. 2013, 25, 391–397. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 17, 78. [Google Scholar] [CrossRef] [Green Version]
- Luschnig, P.; Kienzl, M.; Roula, D.; Pilic, J.; Atallah, R.; Heinemann, A.; Sturm, E.M. The JAK1/2 inhibitor baricitinib suppresses eosinophil effector function and restricts allergen-induced airway eosinophilia. Biochem. Pharmacol. 2021, 192, 114690. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathology Score | Histopathological Lesions in the Brain | Perivascular Cuffing within Meninges | Histopathology in the Nasal Cavity (inc. Mucosa) |
|---|---|---|---|
| 0 | Within normal limits | Within normal limits | Within normal limits |
| 1 | Minimal spongiosis | Minimal | Minimal |
| 2 | Mild spongiosis and minimal to mild increase in neuropil cellularity (glia) | Mild | Mild |
| 3 | Moderate spongiosis and mild neuronal death (as observed within neuronal soma) Moderate increase in neuropil cellularity (glia) | Moderate | Moderate |
| 4 | Moderate to severe spongiosis and neuronal death (as observed within neuronal soma) Moderate increase in neuropil cellularity (glia) | Marked/severe | Marked/severe necrosis of mucosa and presence of exudate within lumen |
| Principal Component | Percentage of the Data Related to Component | Main Associated Components | Correlation to Clinical Signs of Disease? | Interpretation |
|---|---|---|---|---|
| 1 | 26.5% | Th1-biased inflammation markers in the blood and spleen, varying considerably between time points Viral titres in blood and spleen, rapid increase with slow decline | No | Systemic disease, with rapid conventional host response |
| 2 | 18.5% | Th1-biased inflammation markers in the brain, varying considerably between time points Viral titres in brain, rapid increase over time | Yes | A strong association with clinical signs of disease (typically neurological), likely representative of the encephalitis typical of lethal infection |
| 3 | 12.6% | Th2- and Th17-biased inflammation markers in all sample types did not differ between time points | No | Inter-experimental variation in flow cytometry analysis |
| 4 | 7.0% | Cell counts in the blood and spleen did not vary between time points | No | Inter-experimental variation in luminex analysis |
| 5 | 5.3% | Leukocyte counts in the blood, transient decline at the midway point of sampling | No | Leukopenia |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 Crown Copyright. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phelps, A.L.; Salguero, F.J.; Hunter, L.; Stoll, A.L.; Jenner, D.C.; O’Brien, L.M.; Williamson, E.D.; Lever, M.S.; Laws, T.R. Tumour Necrosis Factor-α, Chemokines, and Leukocyte Infiltrate Are Biomarkers for Pathology in the Brains of Venezuelan Equine Encephalitis (VEEV)-Infected Mice. Viruses 2023, 15, 1307. https://doi.org/10.3390/v15061307
Phelps AL, Salguero FJ, Hunter L, Stoll AL, Jenner DC, O’Brien LM, Williamson ED, Lever MS, Laws TR. Tumour Necrosis Factor-α, Chemokines, and Leukocyte Infiltrate Are Biomarkers for Pathology in the Brains of Venezuelan Equine Encephalitis (VEEV)-Infected Mice. Viruses. 2023; 15(6):1307. https://doi.org/10.3390/v15061307
Chicago/Turabian StylePhelps, Amanda L., Francisco J. Salguero, Laura Hunter, Alexander L. Stoll, Dominic C. Jenner, Lyn M. O’Brien, E. Diane Williamson, M. Stephen Lever, and Thomas R. Laws. 2023. "Tumour Necrosis Factor-α, Chemokines, and Leukocyte Infiltrate Are Biomarkers for Pathology in the Brains of Venezuelan Equine Encephalitis (VEEV)-Infected Mice" Viruses 15, no. 6: 1307. https://doi.org/10.3390/v15061307