
ZIKV Strains Elicit Different Inflammatory and Anti-Viral Responses in Microglia Cells

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus and Cells
2.2. BV2 Cells Infection and Treatment
2.3. Cell Viability Assays
2.4. Analysis of BV2 Infection by Immunofluorescence, Quantitative Reverse Transcription PCR (qRT-PCR), and Plaque Assay
2.5. Evaluation of BV2 Cells Activation
2.6. Quantification of microRNAs and Inflammatory Mediator’s Expression in ZIKV-Infected BV2 Cell Line
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
3.1. African Lineage-Derived ZIKVMR766 Strain Infects and Showed Higher Replication Levels in BV2 Microglial Cells Than Asian Lineage-Derived ZIKVPE243 Strain
3.2. African Lineage-Derived ZIKVMR766 Strain Induces Greater BV2 Microglia Cells Activation Than ZIKVPE243
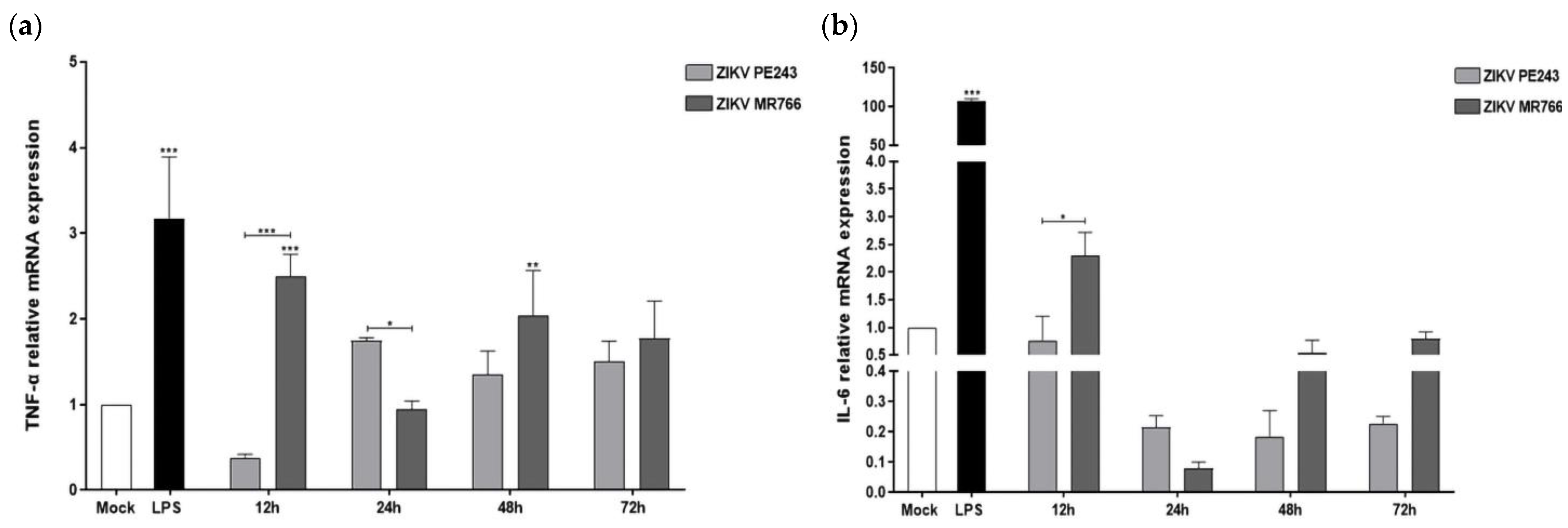
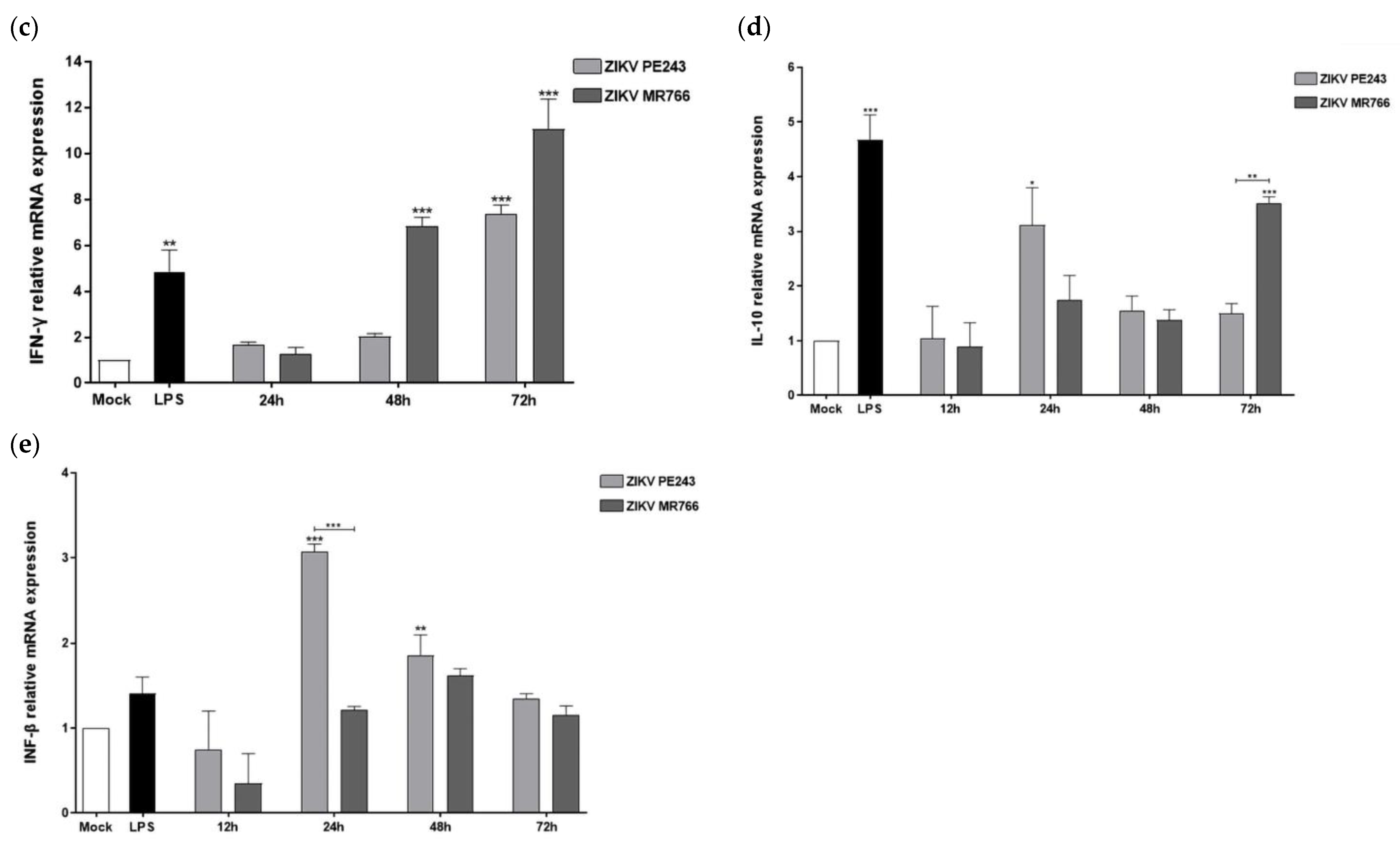
3.3. ZIKVPE243 and ZIKVMR766 Differentially Modulate Pro- and Anti-Inflammatory Cytokines and Anti-Viral Factors’ Expression in BV2 Microglial Cell
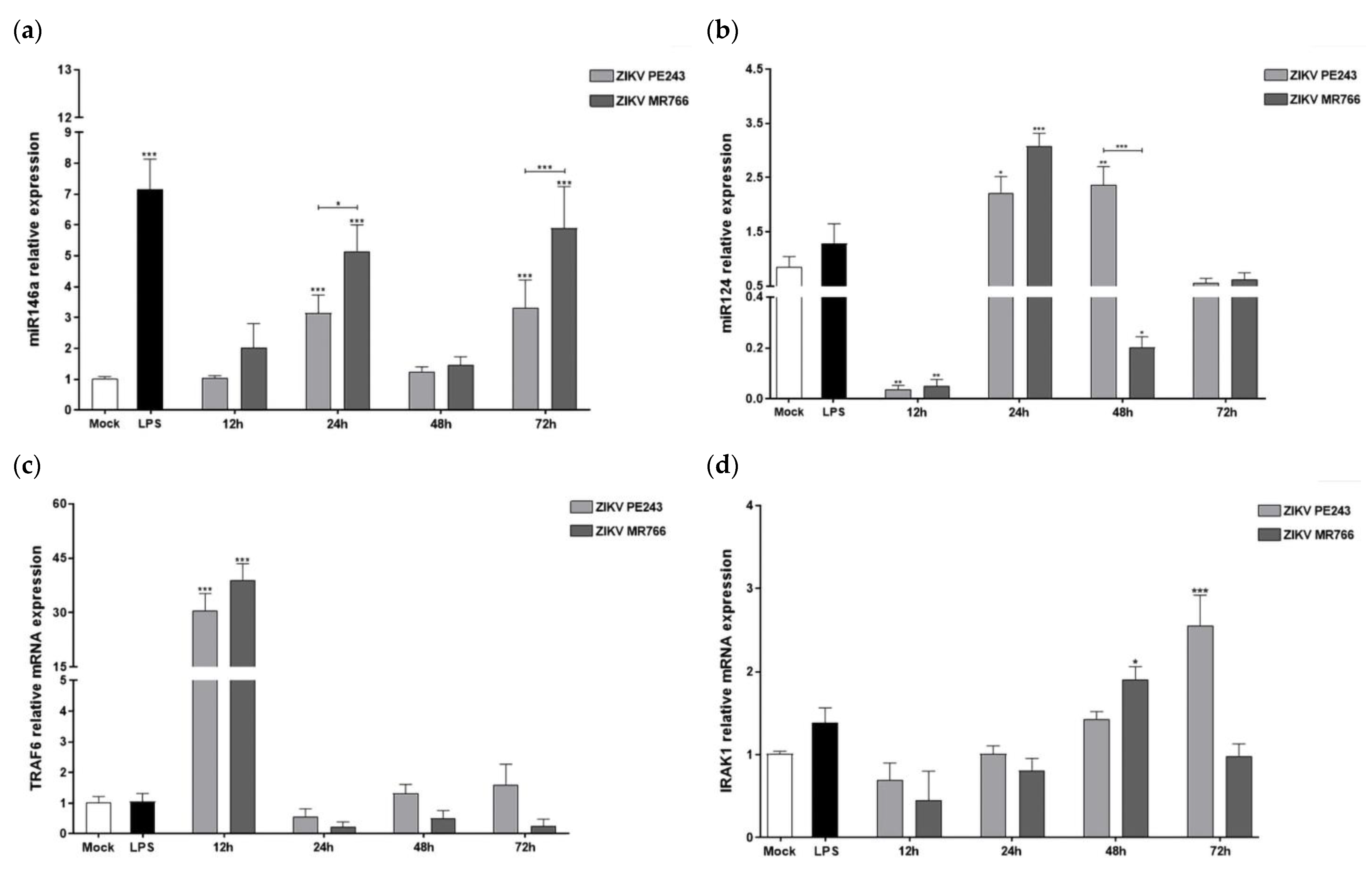
3.4. ZIKVPE243 and ZIKVMR766 Strains Differentially Modulate the Pro-Inflammatory miRNA-155 and Anti-Inflammatory and Anti-Innate Immune Response Factors in BV2 Microglial Cells
3.5. ZIKVPE243 and ZIKVMR766 Differentially Modulate the Anti-Inflammatory miRNAs-146a/124 and Inflammatory Mediators’ Expressions
3.6. BV2 Cells Infected with ZIKVMR766 Display Lower PPAR-γ Expression Levels
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fagbami, A.H. Zika Virus Infections in Nigeria: Virological and Seroepidemiological Investigations in Oyo State. J. Hyg. (Lond.) 1979, 83, 213–219. [Google Scholar] [CrossRef]
- Faye, O.; Faye, O.; de Melo Freire, C.C.; de Oliveira, J.V.; Rubing, C.; de Andrade Zanotto, P.M.; Mawlouth, D.; Sall, A.A. Molecular Evolution of Zika Virus, an Neglected Emerging Disease in Africa and Asia. BMC Proc. 2011, 5, P59. [Google Scholar] [CrossRef]
- Duffy, M.R.; Chen, T.-H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika Virus Outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.-M. Zika Virus, French Polynesia, South Pacific, 2013. Emerg. Infect. Dis. 2014, 20, 1960. [Google Scholar] [CrossRef]
- Zanluca, C.; Melo, V.C.A.d.; Mosimann, A.L.P.; Santos, G.I.V.d.; Santos, C.N.D.d.; Luz, K. First Report of Autochthonous Transmission of Zika Virus in Brazil. Mem. Inst. Oswaldo Cruz 2015, 110, 569–572. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Ishtiaq, F.; Setoh, Y.X.; Ko, A.I. Misperceived Risks of Zika-Related Microcephaly in India. Trends Microbiol. 2019, 27, 381–383. [Google Scholar] [CrossRef]
- Chimelli, L.; Melo, A.S.O.; Avvad-Portari, E.; Wiley, C.A.; Camacho, A.H.S.; Lopes, V.S.; Machado, H.N.; Andrade, C.V.; Dock, D.C.A.; Moreira, M.E.; et al. The Spectrum of Neuropathological Changes Associated with Congenital Zika Virus Infection. Acta Neuropathol. 2017, 133, 983–999. [Google Scholar] [CrossRef]
- del Campo, M.; Feitosa, I.M.L.; Ribeiro, E.M.; Horovitz, D.D.G.; Pessoa, A.L.S.; França, G.V.A.; García-Alix, A.; Doriqui, M.J.R.; Wanderley, H.Y.C.; Sanseverino, M.V.T.; et al. The Phenotypic Spectrum of Congenital Zika Syndrome. Am. J. Med. Genet. Part A 2017, 173, 841–857. [Google Scholar] [CrossRef]
- França, G.V.A.; Schuler-Faccini, L.; Oliveira, W.K.; Henriques, C.M.P.; Carmo, E.H.; Pedi, V.D.; Nunes, M.L.; Castro, M.C.; Serruya, S.; Silveira, M.F.; et al. Congenital Zika Virus Syndrome in Brazil: A Case Series of the First 1501 Livebirths with Complete Investigation. Lancet 2016, 388, 891–897. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.-M.; Blake, A.; Mons, S.; Lastère, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barré Syndrome Outbreak Associated with Zika Virus Infection in French Polynesia: A Case-Control Study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Mécharles, S.; Herrmann, C.; Poullain, P.; Tran, T.-H.; Deschamps, N.; Mathon, G.; Landais, A.; Breurec, S.; Lannuzel, A. Acute Myelitis Due to Zika Virus Infection. Lancet 2016, 387, 1481. [Google Scholar] [CrossRef] [PubMed]
- Reynoso, G.V.; Gordon, D.N.; Kalia, A.; Aguilar, C.C.; Malo, C.S.; Aleshnick, M.; Dowd, K.A.; Cherry, C.R.; Shannon, J.P.; Vrba, S.M.; et al. Zika Virus Spreads through Infection of Lymph Node-Resident Macrophages. bioRxiv 2022, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Driggers, R.W.; Ho, C.-Y.; Korhonen, E.M.; Kuivanen, S.; Jääskeläinen, A.J.; Smura, T.; Rosenberg, A.; Hill, D.A.; DeBiasi, R.L.; Vezina, G.; et al. Zika Virus Infection with Prolonged Maternal Viremia and Fetal Brain Abnormalities. N. Engl. J. Med. 2016, 374, 2142–2151. [Google Scholar] [CrossRef]
- Papa, M.P.; Meuren, L.M.; Coelho, S.V.A.; Lucas, C.G.d.O.; Mustafá, Y.M.; Lemos Matassoli, F.; Silveira, P.P.; Frost, P.S.; Pezzuto, P.; Ribeiro, M.R.; et al. Zika Virus Infects, Activates, and Crosses Brain Microvascular Endothelial Cells, without Barrier Disruption. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Simonin, Y.; van Riel, D.; Van de Perre, P.; Rockx, B.; Salinas, S. Differential Virulence between Asian and African Lineages of Zika Virus. PLoS Negl. Trop. Dis. 2017, 11, e0005821. [Google Scholar] [CrossRef]
- Alwis, R.d.; Zellweger, R.M.; Chua, E.; Wang, L.-F.; Chawla, T.; Sessions, O.M.; Marlier, D.; Connolly, J.; Messling, V.v.; Anderson, D.E. Systemic Inflammation, Innate Immunity and Pathogenesis after Zika Virus Infection in Cynomolgus Macaques Are Modulated by Strain-Specificity within the Asian Lineage. Emerg. Microbes Infect. 2021, 1–31. [Google Scholar] [CrossRef]
- Dowall, S.D.; Graham, V.A.; Rayner, E.; Atkinson, B.; Hall, G.; Watson, R.J.; Bosworth, A.; Bonney, L.C.; Kitchen, S.; Hewson, R. A Susceptible Mouse Model for Zika Virus Infection. PLoS Negl. Trop. Dis. 2016, 10, e0004658. [Google Scholar] [CrossRef]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef]
- Esser-Nobis, K.; Aarreberg, L.D.; Roby, J.A.; Fairgrieve, M.R.; Green, R.; Gale, M. Comparative Analysis of African and Asian Lineage-Derived Zika Virus Strains Reveals Differences in Activation of and Sensitivity to Antiviral Innate Immunity. J. Virol. 2019, 93, 1–18. [Google Scholar] [CrossRef]
- Lucas, C.G.O.; Kitoko, J.Z.; Ferreira, F.M.; Suzart, V.G.; Papa, M.P.; Coelho, S.V.A.; Cavazzoni, C.B.; Paula-Neto, H.A.; Olsen, P.C.; Iwasaki, A.; et al. Critical Role of CD4+ T Cells and IFNγ Signaling in Antibody-Mediated Resistance to Zika Virus Infection. Nat. Commun. 2018, 9, 3136. [Google Scholar] [CrossRef]
- Yang, S.; Gorshkov, K.; Lee, E.M.; Xu, M.; Cheng, Y.-S.; Sun, N.; Soheilian, F.; de Val, N.; Ming, G.; Song, H.; et al. Zika Virus-Induced Neuronal Apoptosis via Increased Mitochondrial Fragmentation. Front. Microbiol. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- de Sousa, J.R.; do Socorro da Silva Azevedo, R.; Quaresma, J.A.S.; da Costa Vasconcelos, P.F. The Innate Immune Response in Zika Virus Infection. Rev. Med. Virol. 2021, 31, e2166. [Google Scholar] [CrossRef]
- Chazal, M.; Beauclair, G.; Gracias, S.; Najburg, V.; Simon-Lorière, E.; Tangy, F.; Komarova, A.V.; Jouvenet, N. RIG-I Recognizes the 5′ Region of Dengue and Zika Virus Genomes. Cell Rep. 2018, 24, 320–328. [Google Scholar] [CrossRef]
- Sprokholt, J.K.; Kaptein, T.M.; van Hamme, J.L.; Overmars, R.J.; Gringhuis, S.I.; Geijtenbeek, T.B.H. RIG-I-like Receptor Activation by Dengue Virus Drives Follicular T Helper Cell Formation and Antibody Production. PLOS Pathog. 2017, 13, e1006738. [Google Scholar] [CrossRef]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef]
- Nazmi, A.; Dutta, K.; Hazra, B.; Basu, A. Role of Pattern Recognition Receptors in Flavivirus Infections. Virus Res. 2014, 185, 32–40. [Google Scholar] [CrossRef]
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.; Bramley, J.C.; Morosky, S.; Marques, E.T.D.A.; Cherry, S.; Sadovsky, Y.; Coyne, C.B. Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe 2016, 19, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.R.; Quicke, K.M.; Maddur, M.S.; O’Neal, J.T.; McDonald, C.E.; Fedorova, N.B.; Puri, V.; Shabman, R.S.; Pulendran, B.; Suthar, M.S. Zika Virus Antagonizes Type I Interferon Responses during Infection of Human Dendritic Cells. PLOS Pathog. 2017, 13, e1006164. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika Virus Inhibits Type-I Interferon Production and Downstream Signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sánchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Sheedy, F.J.; McCoy, C.E. MicroRNAs: The Fine-Tuners of Toll-like Receptor Signalling. Nat. Rev. Immunol. 2011, 11, 163–175. [Google Scholar] [CrossRef]
- Su, W.; Aloi, M.S.; Garden, G.A. MicroRNAs Mediating CNS Inflammation: Small Regulators with Powerful Potential. Brain. Behav. Immun. 2016, 52, 1–8. [Google Scholar] [CrossRef]
- Kumar, M.; Nerurkar, V.R. Integrated Analysis of MicroRNAs and Their Disease Related Targets in the Brain of Mice Infected with West Nile Virus. Virology 2014, 452–453, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popović, M.; Poljšak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodušek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef]
- Jorgačevski, J.; Korva, M.; Potokar, M.; Lisjak, M.; Avšič-Županc, T.; Zorec, R. ZIKV Strains Differentially Affect Survival of Human Fetal Astrocytes versus Neurons and Traffic of ZIKV-Laden Endocytotic Compartments. Sci. Rep. 2019, 9, 8069. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative Activation of Macrophages: Mechanism and Functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Angeles Muñoz-Fernández, M.; Fresno, M. The Role of Tumour Necrosis Factor, Interleukin 6, Interferon-γ and Inducible Nitric Oxide Synthase in the Development and Pathology of the Nervous System. Prog. Neurobiol. 1998, 56, 307–340. [Google Scholar] [CrossRef] [PubMed]
- Lum, F.-M.; Low, D.K.S.; Fan, Y.; Tan, J.J.L.; Lee, B.; Chan, J.K.Y.; Rénia, L.; Ginhoux, F.; Ng, L.F.P. Zika Virus Infects Human Fetal Brain Microglia and Induces Inflammation. Clin. Infect. Dis. 2017, 64, 914–920. [Google Scholar] [CrossRef]
- Saijo, K.; Crotti, A.; Glass, C.K. Regulation of Microglia Activation and Deactivation by Nuclear Receptors. Glia 2013, 61, 104–111. [Google Scholar] [CrossRef]
- Zolezzi, J.M.; Santos, M.J.; Bastías-Candia, S.; Pinto, C.; Godoy, J.A.; Inestrosa, N.C. PPARs in the Central Nervous System: Roles in Neurodegeneration and Neuroinflammation. Biol. Rev. 2017, 92, 2046–2069. [Google Scholar] [CrossRef]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef] [PubMed]
- Li-hua, D.; Yan, L.; Shi-ji, W.; Guang, W.; Lu-lu, S.; Xue-feng, P.; Pengda, S. Esculentoside A Inhibits LPS-Induced BV2 Microglia Activation through Activating PPAR-γ. Eur. J. Pharmacol. 2017, 813, 61–65. [Google Scholar] [CrossRef]
- Pisanu, A.; Lecca, D.; Mulas, G.; Wardas, J.; Simbula, G.; Spiga, S.; Carta, A.R. Dynamic Changes in Pro- and Anti-Inflammatory Cytokines in Microglia after PPAR-γ Agonist Neuroprotective Treatment in the MPTPp Mouse Model of Progressive Parkinson’s Disease. Neurobiol. Dis. 2014, 71, 280–291. [Google Scholar] [CrossRef]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.Y.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes Are Signaling Platforms for Antiviral Innate Immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef]
- Martín-Acebes, M.A.; Vázquez-Calvo, Á.; Saiz, J.-C. Lipids and Flaviviruses, Present and Future Perspectives for the Control of Dengue, Zika, and West Nile Viruses. Prog. Lipid Res. 2016, 64, 123–137. [Google Scholar] [CrossRef]
- Coelho, S.V.A.; Neris, R.L.S.; Papa, M.P.; Schnellrath, L.C.; Meuren, L.M.; Tschoeke, D.A.; Leomil, L.; Verçoza, B.R.F.; Miranda, M.; Thompson, F.L.; et al. Development of Standard Methods for Zika Virus Propagation, Titration, and Purification. J. Virol. Methods 2017, 246, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Velez, J.O.; Lambert, A.J.; Johnson, A.J.; Stanfield, S.M.; Duffy, M.R. Genetic and Serologic Properties of Zika Virus Associated with an Epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 2008, 14, 1232–1239. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, E.D.; Veremeyko, T.; Barteneva, N.; Krichevsky, A.M.; Weiner, H.L. MicroRNA-124 Promotes Microglia Quiescence and Suppresses EAE by Deactivating Macrophages via the C/EBP-α–PU.1 Pathway. Nat. Med. 2011, 17, 64–70. [Google Scholar] [CrossRef]
- Li, Y.Y.; Cui, J.G.; Dua, P.; Pogue, A.I.; Bhattacharjee, S.; Lukiw, W.J. Differential Expression of MiRNA-146a-Regulated Inflammatory Genes in Human Primary Neural, Astroglial and Microglial Cells. Neurosci. Lett. 2011, 499, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wang, P.; Lin, L.; Liu, X.; Ma, F.; An, H.; Wang, Z.; Cao, X. MicroRNA-146a Feedback Inhibits RIG-I-Dependent Type I IFN Production in Macrophages by Targeting TRAF6, IRAK1, and IRAK2. J. Immunol. 2009, 183, 2150–2158. [Google Scholar] [CrossRef] [PubMed]
- Diop, F.; Vial, T.; Ferraris, P.; Wichit, S.; Bengue, M.; Hamel, R.; Talignani, L.; Liegeois, F.; Pompon, J.; Yssel, H.; et al. Zika Virus Infection Modulates the Metabolomic Profile of Microglial Cells. PLoS ONE 2018, 13, e0206093. [Google Scholar] [CrossRef] [PubMed]
- Carteaux, G.; Maquart, M.; Bedet, A.; Contou, D.; Brugières, P.; Fourati, S.; Cleret de Langavant, L.; de Broucker, T.; Brun-Buisson, C.; Leparc-Goffart, I.; et al. Zika Virus Associated with Meningoencephalitis. N. Engl. J. Med. 2016, 374, 1595–1596. [Google Scholar] [CrossRef]
- Figueiredo, C.P.; Barros-Aragão, F.G.Q.; Neris, R.L.S.; Frost, P.S.; Soares, C.; Souza, I.N.O.; Zeidler, J.D.; Zamberlan, D.C.; de Sousa, V.L.; Souza, A.S.; et al. Zika Virus Replicates in Adult Human Brain Tissue and Impairs Synapses and Memory in Mice. Nat. Commun. 2019, 10, 3890. [Google Scholar] [CrossRef]
- Wang, B.; Thurmond, S.; Zhou, K.; Sánchez-Aparicio, M.T.; Fang, J.; Lu, J.; Gao, L.; Ren, W.; Cui, Y.; Veit, E.C.; et al. Structural Basis for STAT2 Suppression by Flavivirus NS5. Nat. Struct. Mol. Biol. 2020, 27, 875–885. [Google Scholar] [CrossRef]
- Anfasa, F.; Siegers, J.Y.; van der Kroeg, M.; Mumtaz, N.; Stalin Raj, V.; de Vrij, F.M.S.; Widagdo, W.; Gabriel, G.; Salinas, S.; Simonin, Y.; et al. Phenotypic Differences between Asian and African Lineage Zika Viruses in Human Neural Progenitor Cells. mSphere 2017, 2, 1–10. [Google Scholar] [CrossRef]
- Nakayama, E.; Kato, F.; Tajima, S.; Ogawa, S.; Yan, K.; Takahashi, K.; Sato, Y.; Suzuki, T.; Kawai, Y.; Inagaki, T.; et al. Neuroinvasiveness of the MR766 Strain of Zika Virus in IFNAR-/- Mice Maps to PrM Residues Conserved amongst African Genotype Viruses. PLOS Pathog. 2021, 17, e1009788. [Google Scholar] [CrossRef]
- Hendrickx, D.A.E.; van Eden, C.G.; Schuurman, K.G.; Hamann, J.; Huitinga, I. Staining of HLA-DR, Iba1 and CD68 in Human Microglia Reveals Partially Overlapping Expression Depending on Cellular Morphology and Pathology. J. Neuroimmunol. 2017, 309, 12–22. [Google Scholar] [CrossRef]
- Hertzog, J.; Dias Junior, A.G.; Rigby, R.E.; Donald, C.L.; Mayer, A.; Sezgin, E.; Song, C.; Jin, B.; Hublitz, P.; Eggeling, C.; et al. Infection with a Brazilian Isolate of Zika Virus Generates RIG-I Stimulatory RNA and the Viral NS5 Protein Blocks Type I IFN Induction and Signaling. Eur. J. Immunol. 2018, 48, 1120–1136. [Google Scholar] [CrossRef]
- McCoy, C.E.; Sheedy, F.J.; Qualls, J.E.; Doyle, S.L.; Quinn, S.R.; Murray, P.J.; O’Neill, L.A.J. IL-10 Inhibits MiR-155 Induction by Toll-like Receptors. J. Biol. Chem. 2010, 285, 20492–20498. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yang, S.; He, J.; Guest, J.D.; Ma, Z.; Yang, L.; Pierce, B.G.; Tang, Q.; Zhang, Y.-J. Zika Virus NS5 Protein Antagonizes Type I Interferon Production via Blocking TBK1 Activation. Virology 2019, 527, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-Lambda (IFN-λ) Is Expressed in a Tissue-Dependent Fashion and Primarily Acts on Epithelial Cells In Vivo. PLoS Pathog. 2008, 4, e1000017. [Google Scholar] [CrossRef]
- Akhtar, L.N.; Qin, H.; Muldowney, M.T.; Yanagisawa, L.L.; Kutsch, O.; Clements, J.E.; Benveniste, E.N. Suppressor of Cytokine Signaling 3 Inhibits Antiviral IFN-β Signaling To Enhance HIV-1 Replication in Macrophages. J. Immunol. 2010, 185, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, L.N.; Benveniste, E.N. Viral Exploitation of Host SOCS Protein Functions. J. Virol. 2011, 85, 1912–1921. [Google Scholar] [CrossRef]
- Seong, R.-K.; Lee, J.K.; Shin, O.S. Zika Virus-Induction of the Suppressor of Cytokine Signaling 1/3 Contributes to the Modulation of Viral Replication. Pathogens 2020, 9, 163. [Google Scholar] [CrossRef]
- Thounaojam, M.C.; Kundu, K.; Kaushik, D.K.; Swaroop, S.; Mahadevan, A.; Shankar, S.K.; Basu, A. MicroRNA 155 Regulates Japanese Encephalitis Virus-Induced Inflammatory Response by Targeting Src Homology 2-Containing Inositol Phosphatase 1. J. Virol. 2014, 88, 4798–4810. [Google Scholar] [CrossRef]
- Ghoshal, A.; Das, S.; Ghosh, S.; Mishra, M.K.; Sharma, V.; Koli, P.; Sen, E.; Basu, A. Proinflammatory Mediators Released by Activated Microglia Induces Neuronal Death in Japanese Encephalitis. Glia 2007, 55, 483–496. [Google Scholar] [CrossRef]
- Li, X.; Zhu, Q.; Cao, Q.; Chen, H.; Qian, P. Japanese Encephalitis Virus Upregulates the Expression of SOCS3 in Mouse Brain and Raw264.7 Cells. Viruses 2014, 6, 4280–4293. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Sheng, C.; Gao, S.; Yao, C.; Li, J.; Jiang, W.; Chen, H.; Wu, J.; Pan, C.; Chen, S.; et al. SOCS3 Drives Proteasomal Degradation of TBK1 and Negatively Regulates Antiviral Innate Immunity. Mol. Cell. Biol. 2015, 35, 2400–2413. [Google Scholar] [CrossRef]
- Blumer, T.; Coto-Llerena, M.; Duong, F.H.T.; Heim, M.H. SOCS1 Is an Inducible Negative Regulator of Interferon λ (IFN-λ)–Induced Gene Expression in Vivo. J. Biol. Chem. 2017, 292, 17928–17938. [Google Scholar] [CrossRef] [PubMed]
- Alexander, W.S.; Starr, R.; Fenner, J.E.; Scott, C.L.; Handman, E.; Sprigg, N.S.; Corbin, J.E.; Cornish, A.L.; Darwiche, R.; Owczarek, C.M.; et al. SOCS1 Is a Critical Inhibitor of Interferon γ Signaling and Prevents the Potentially Fatal Neonatal Actions of This Cytokine. Cell 1999, 98, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Ubol, S.; Phuklia, W.; Kalayanarooj, S.; Modhiran, N. Mechanisms of Immune Evasion Induced by a Complex of Dengue Virus and Preexisting Enhancing Antibodies. J. Infect. Dis. 2010, 201, 923–935. [Google Scholar] [CrossRef]
- Flores-Mendoza, L.K.; Estrada-Jiménez, T.; Sedeño-Monge, V.; Moreno, M.; Manjarrez, M.D.C.; González-Ochoa, G.; Millán-Pérez Peña, L.; Reyes-Leyva, J. IL-10 and Socs3 Are Predictive Biomarkers of Dengue Hemorrhagic Fever. Mediators Inflamm. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Kapadia, R. Mechanisms of Anti-Inflammatory and Neuroprotective Actions of PPAR-Gamma Agonists. Front. Biosci. 2008, 13, 1813. [Google Scholar] [CrossRef] [PubMed]
- Foo, S.; Chen, W.; Chan, Y.; Bowman, J.W.; Chang, L.; Choi, Y.; Yoo, J.S.; Ge, J.; Cheng, G.; Bonnin, A.; et al. Asian Zika Virus Strains Target CD14+ Blood Monocytes and Induce M2-Skewed Immunosuppression during Pregnancy. Nat. Microbiol. 2017, 2, 1558–1570. [Google Scholar] [CrossRef]
- Viladomiu, M.; Hontecillas, R.; Pedragosa, M.; Carbo, A.; Hoops, S.; Michalak, P.; Michalak, K.; Guerrant, R.L.; Roche, J.K.; Warren, C.A.; et al. Modeling the Role of Peroxisome Proliferator-Activated Receptor γ and MicroRNA-146 in Mucosal Immune Responses to Clostridium Difficile. PLoS ONE 2012, 7, e47525. [Google Scholar] [CrossRef]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef]
- Thulasi Raman, S.N.; Latreille, E.; Gao, J.; Zhang, W.; Wu, J.; Russell, M.S.; Walrond, L.; Cyr, T.; Lavoie, J.R.; Safronetz, D.; et al. Dysregulation of Ephrin Receptor and PPAR Signaling Pathways in Neural Progenitor Cells Infected by Zika Virus. Emerg. Microbes Infect. 2020, 9, 2046–2060. [Google Scholar] [CrossRef]
- You, J.; Hou, S.; Malik-Soni, N.; Xu, Z.; Kumar, A.; Rachubinski, R.A.; Frappier, L.; Hobman, T.C. Flavivirus Infection Impairs Peroxisome Biogenesis and Early Antiviral Signaling. J. Virol. 2015, 89, 12349–12361. [Google Scholar] [CrossRef]
- Coyaud, E.; Ranadheera, C.; Cheng, D.; Gonçalves, J.; Dyakov, B.J.A.; Laurent, E.M.N.; St-Germain, J.; Pelletier, L.; Gingras, A.-C.; Brumell, J.H.; et al. Global Interactomics Uncovers Extensive Organellar Targeting by Zika Virus. Mol. Cell. Proteomics 2018, 17, 2242–2255. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, A.; Pinto, I.F.D.; Lima, M.; Giovanetti, M.; de Jesus, J.G.; Xavier, J.; Barreto, F.K.; Canuto, G.A.B.; do Amaral, H.R.; de Filippis, A.M.B.; et al. Lipidomic Analysis Reveals Serum Alteration of Plasmalogens in Patients Infected With ZIKA Virus. Front. Microbiol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wei, C.; Xiao, X.; Dong, Y.; Tan, B.; Yu, J.; Chen, G.; Yuan, Q.; Du, Z.; Sun, Y.; et al. Expression of TNF-α and IL-β Can Be Suppressed via the PPAR-γ/MTOR Signaling Pathway in BV-2 Microglia: A Potential Anti-inflammation Mechanism. Mol. Med. Rep. 2020, 22, 3559–3565. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.-S.; Lee, S.-A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-MTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef]
- Akiyama, B.M.; Laurence, H.M.; Massey, A.R.; Costantino, D.A.; Xie, X.; Yang, Y.; Shi, P.-Y.; Nix, J.C.; Beckham, J.D.; Kieft, J.S. Zika Virus Produces Noncoding RNAs Using a Multi-Pseudoknot Structure That Confounds a Cellular Exonuclease. Science 2016, 354, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Donald, C.L.; Brennan, B.; Cumberworth, S.L.; Rezelj, V.V.; Clark, J.J.; Cordeiro, M.T.; Freitas de Oliveira França, R.; Pena, L.J.; Wilkie, G.S.; Da Silva Filipe, A.; et al. Full Genome Sequence and SfRNA Interferon Antagonist Activity of Zika Virus from Recife, Brazil. PLoS Negl. Trop. Dis. 2016, 10, e0005048. [Google Scholar] [CrossRef]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue Subgenomic RNA Binds TRIM25 to Inhibit Interferon Expression for Epidemiological Fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef]
- Schnettler, E.; Sterken, M.G.; Leung, J.Y.; Metz, S.W.; Geertsema, C.; Goldbach, R.W.; Vlak, J.M.; Kohl, A.; Khromykh, A.A.; Pijlman, G.P. Noncoding Flavivirus RNA Displays RNA Interference Suppressor Activity in Insect and Mammalian Cells. J. Virol. 2012, 86, 13486–13500. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 12 hpi | 24 hpi | 48 hpi | 72 hpi | |||||
|---|---|---|---|---|---|---|---|---|
| PE243 | MR766 | PE243 | MR766 | PE243 | MR766 | PE243 | MR766 | |
| Inflammatory Markers | ||||||||
| TNF-α | 0.37 | 2.49 | 1.75 | 0.95 | 1.35 | 2.04 | 1.50 | 1.77 |
| IL-6 | 0.77 | 2.30 | 0.22 | 0.08 | 0.18 | 0.55 | 0.23 | 0.80 |
| IFN-γ | - | - | 1.68 | 1.26 | 2.03 | 6.83 | 7.36 | 11.05 |
| miRNA155 | 1.02 | 1.63 | 1.81 | 2.14 | 0.92 | 1.05 | 4.10 | 2.41 |
| TRAF6 | 30.02 | 38.23 | 0.55 | 0.22 | 1.30 | 0.48 | 1.56 | 0.25 |
| IRAK1 | 0.69 | 0.45 | 1.00 | 0.80 | 1.42 | 1.89 | 2.53 | 0.97 |
| Anti-Inflammatory Markers | ||||||||
| IL-10 | 1.04 | 0.89 | 3.12 | 1.75 | 1.55 | 1.38 | 1.50 | 3.52 |
| PPAR-γ | 5.53 | 2.84 | 11.05 | 0.37 | 24.65 | 5.15 | 30.13 | 26.33 |
| miRNA146a | 1.01 | 1.98 | 3.04 | 4.99 | 1.21 | 1.41 | 3.23 | 5.72 |
| miRNA124 | 0.04 | 0.06 | 2.63 | 3.65 | 2.80 | 0.24 | 0.66 | 0.73 |
| SHIP1 | 8.98 | 4.39 | 2.44 | 0.38 | 0.23 | 0.14 | 0.33 | 0.24 |
| Anti-Viral Response | ||||||||
| SOCS1 | 20.68 | 7.34 | 4.51 | 1.17 | 0.27 | 0.23 | 0.14 | 0.06 |
| SOCS3 | 0.40 | 0.46 | 0.88 | 0.92 | 1.17 | 1.76 | 1.28 | 1.82 |
| IFN-β | 0.75 | 0.35 | 3.07 | 1.22 | 1.86 | 1.62 | 1.34 | 1.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, F.B.C.d.; Freire, V.P.A.S.d.S.; Coelho, S.V.A.; Meuren, L.M.; Palmeira, J.d.F.; Cardoso, A.L.; Neves, F.d.A.R.; Ribeiro, B.M.; Argañaraz, G.A.; Arruda, L.B.d.; et al. ZIKV Strains Elicit Different Inflammatory and Anti-Viral Responses in Microglia Cells. Viruses 2023, 15, 1250. https://doi.org/10.3390/v15061250
Oliveira FBCd, Freire VPASdS, Coelho SVA, Meuren LM, Palmeira JdF, Cardoso AL, Neves FdAR, Ribeiro BM, Argañaraz GA, Arruda LBd, et al. ZIKV Strains Elicit Different Inflammatory and Anti-Viral Responses in Microglia Cells. Viruses. 2023; 15(6):1250. https://doi.org/10.3390/v15061250
Chicago/Turabian StyleOliveira, Fernanda Bellaniza Caminha de, Vanessa Paola Alves Sampaio de Sá Freire, Sharton Vinicius Antunes Coelho, Lana Monteiro Meuren, Julys da Fonseca Palmeira, Ana Luísa Cardoso, Francisco de Assis Rocha Neves, Bergmann Morais Ribeiro, Gustavo Adolfo Argañaraz, Luciana Barros de Arruda, and et al. 2023. "ZIKV Strains Elicit Different Inflammatory and Anti-Viral Responses in Microglia Cells" Viruses 15, no. 6: 1250. https://doi.org/10.3390/v15061250