
Temsavir Modulates HIV-1 Envelope Conformation by Decreasing Its Proteolytic Cleavage
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cell Lines and Isolation of Primary Cells
2.3. Plasmids and Proviral Constructs
2.4. Viral Production and Infections
2.5. Antibodies and HIV+ Plasma
2.6. Small Molecules
2.7. Radioactive Labeling and Immunoprecipitation of Envelope Glycoproteins
2.8. Transfection
2.9. Virus Neutralization Assay
2.10. Flow Cytometry Analysis of Cell-Surface Staining
2.11. FACS-Based ADCC Assay
2.12. Detection of Soluble gp120 by Sandwich ELISA
2.13. Statistical Analysis
3. Results
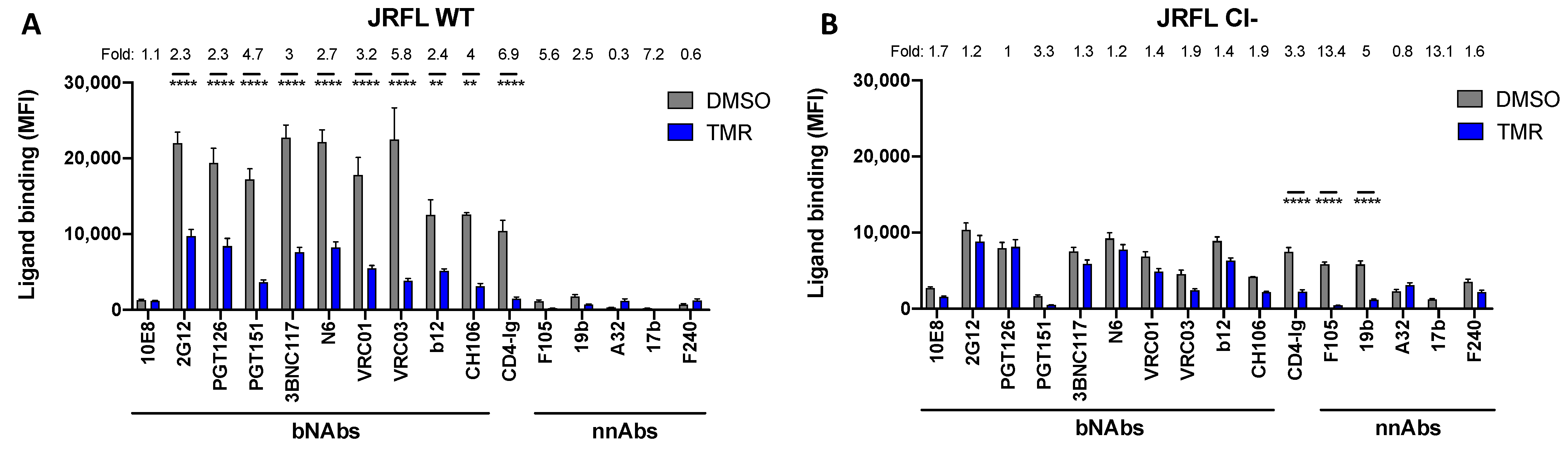
3.1. The Effect of Temsavir on Env Depends on Proteolytic Env Processing
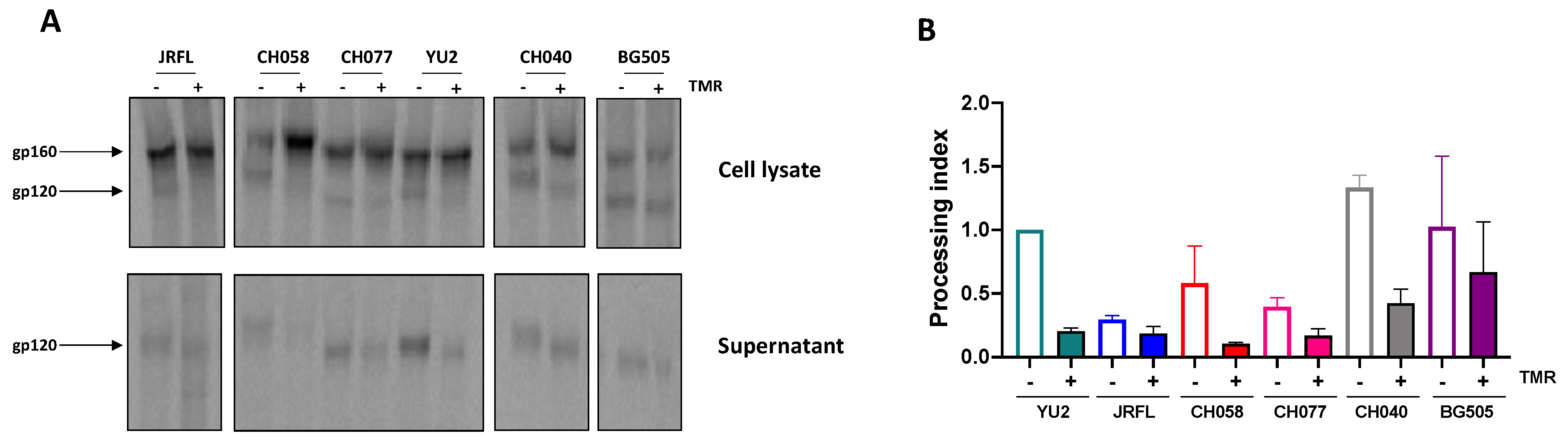
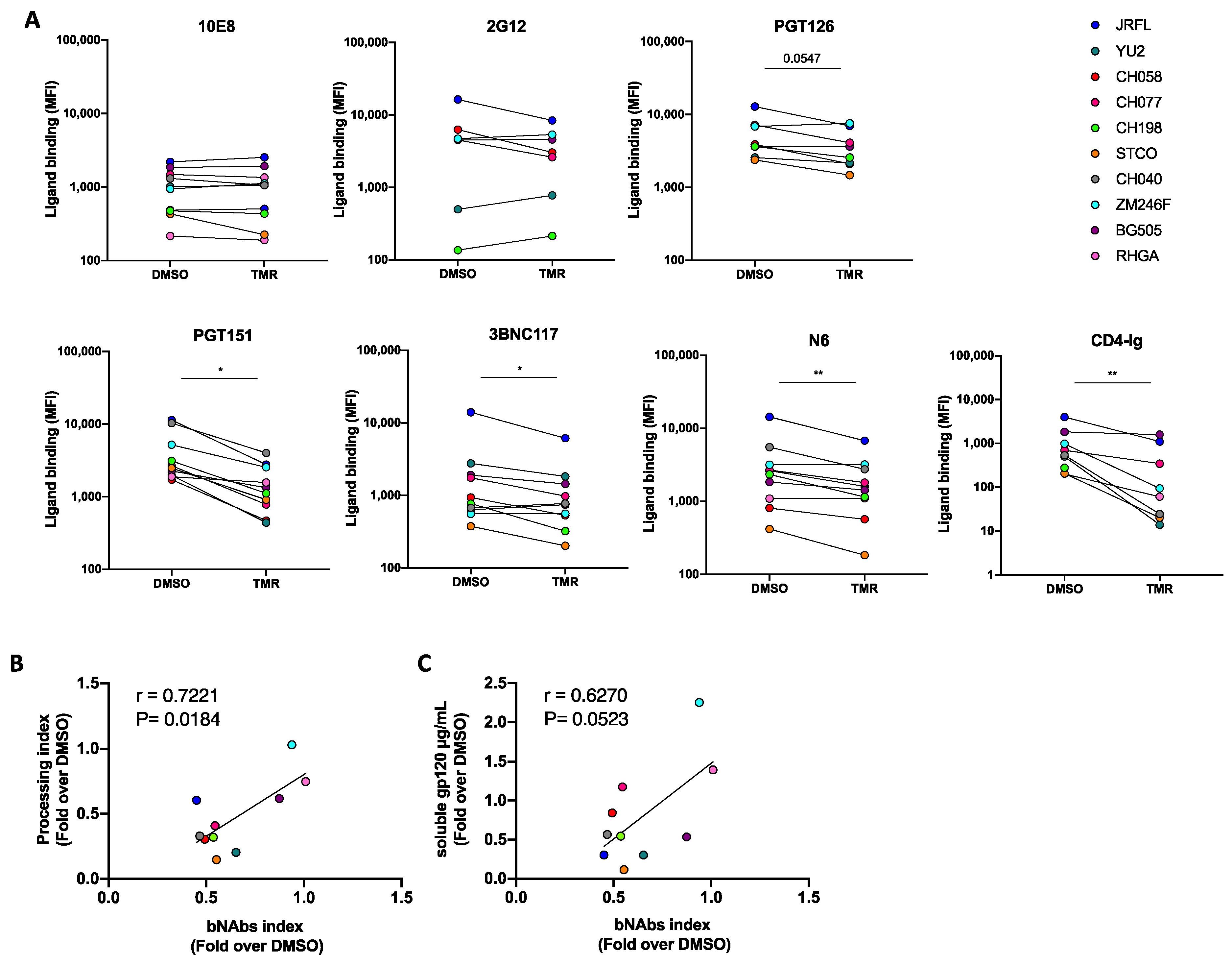
3.2. Temsavir Decreases Glycosylation and the Proteolytic Cleavage of Env, Resulting in Decreased Recognition by bNAbs
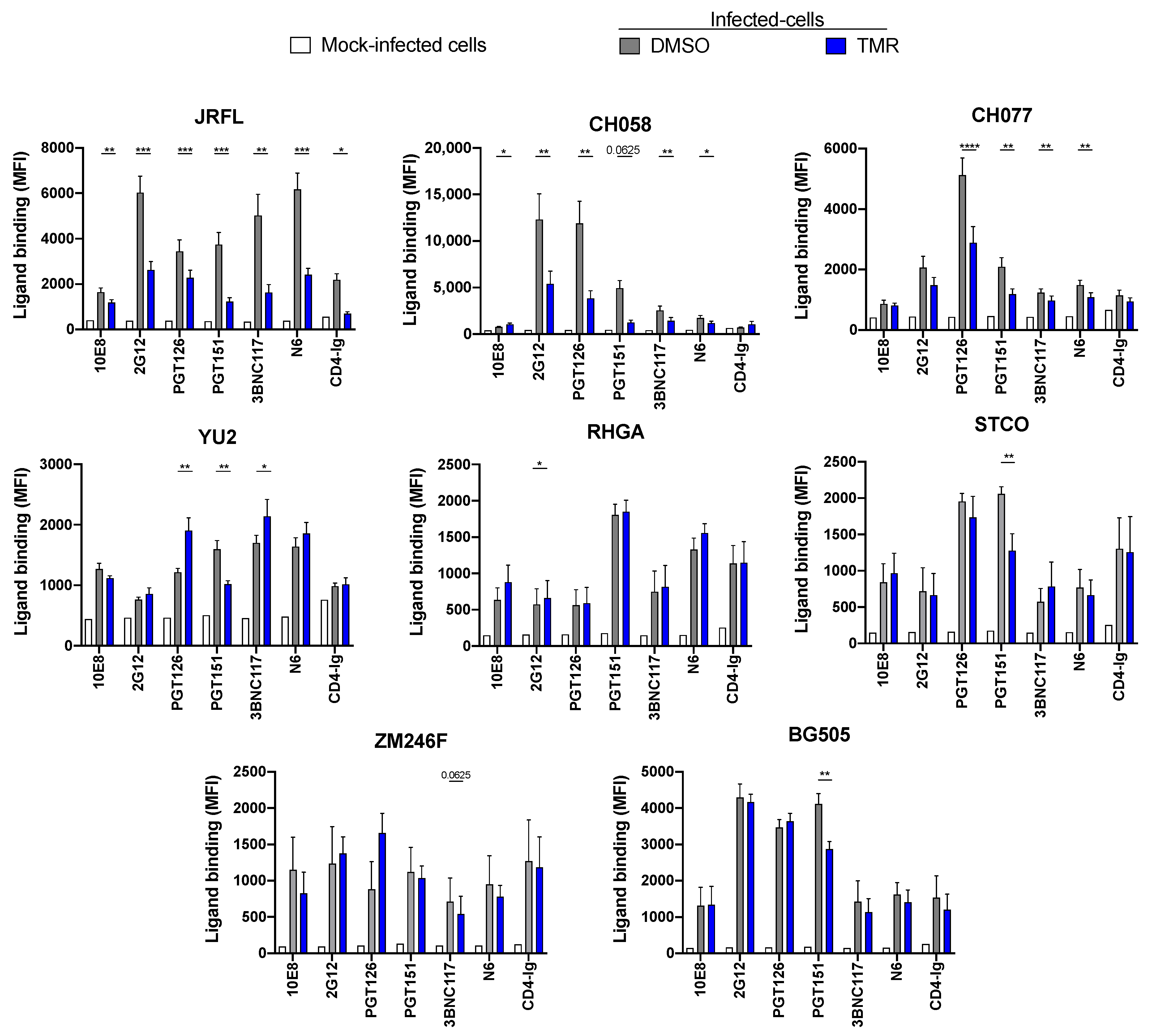
3.3. Temsavir Affects Env Recognition of Infected Primary CD4+ T Cells
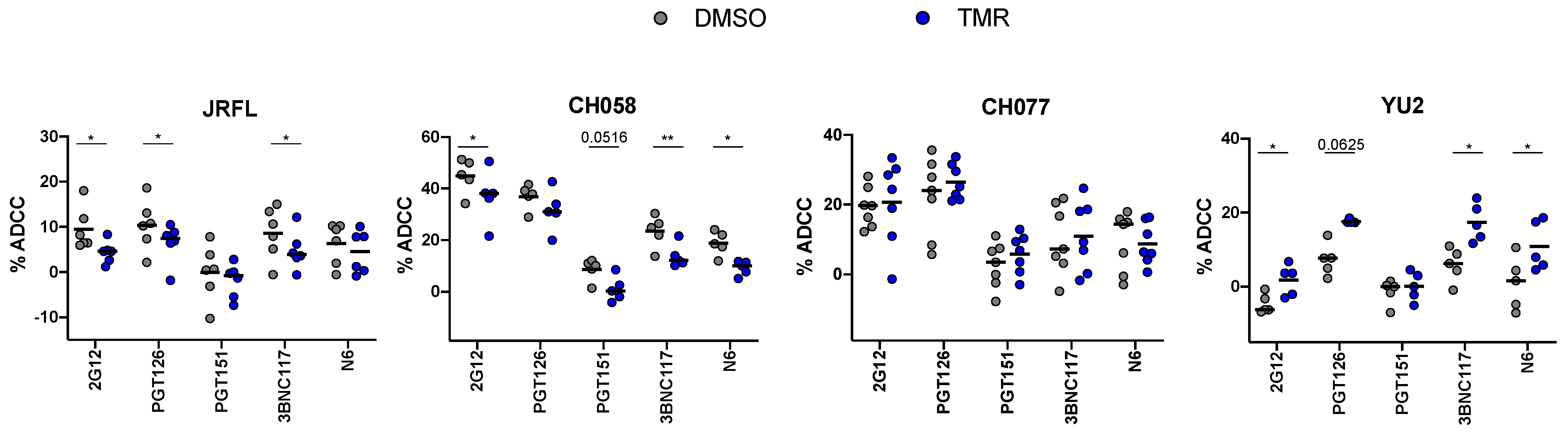
3.4. Impact of Temsavir on bNAb Recognition Affects ADCC Responses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Center, R.J.; Leapman, R.D.; Lebowitz, J.; Arthur, L.O.; Earl, P.L.; Moss, B. Oligomeric structure of the human immunodeficiency virus type 1 envelope protein on the virion surface. J. Virol. 2002, 76, 7863–7867. [Google Scholar] [CrossRef] [PubMed]
- McCune, J.M.; Rabin, L.B.; Feinberg, M.B.; Lieberman, M.; Kosek, J.C.; Reyes, G.R.; Weissman, I.L. Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell 1988, 53, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.; Myers, D.; Risser, R. Mutational analysis of the cleavage sequence of the human immunodeficiency virus type 1 envelope glycoprotein precursor gp160. J. Virol. 1989, 63, 4670–4675. [Google Scholar] [CrossRef] [PubMed]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell binding and entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Ma, X.; Castillo-Menendez, L.R.; Gorman, J.; Alsahafi, N.; Ermel, U.; Terry, D.S.; Chambers, M.; Peng, D.; Zhang, B.; et al. Associating HIV-1 envelope glycoprotein structures with states on the virus observed by smFRET. Nature 2019, 568, 415–419. [Google Scholar] [CrossRef]
- Li, Z.; Li, W.; Lu, M.; Bess, J.; Chao, C.W.; Gorman, J.; Terry, D.S.; Zhang, B.; Zhou, T.; Blanchard, S.C.; et al. Subnanometer structures of HIV-1 envelope trimers on aldrithiol-2-inactivated virus particles. Nat. Struct. Mol. Biol. 2020, 27, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Gorman, J.; Ma, X.; Zhou, Z.; Arthos, J.; Burton, D.R.; Koff, W.C.; Courter, J.R.; Smith, A.B., III; Kwong, P.D.; et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 2014, 346, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Stadtmueller, B.M.; Bridges, M.D.; Dam, K.-M.; Lerch, M.T.; Huey-Tubman, K.E.; Hubbell, W.L.; Bjorkman, P.J. DEER spectroscopy measurements reveal multiple conformations of HIV-1 SOSIP envelopes that show similarities with envelopes on native virions. Immunity 2018, 49, 235–246.e4. [Google Scholar] [CrossRef]
- Alsahafi, N.; Bakouche, N.; Kazemi, M.; Richard, J.; Ding, S.; Bhattacharyya, S.; Das, D.; Anand, S.P.; Prévost, J.; Tolbert, W.D.; et al. An Asymmetric Opening of HIV-1 Envelope Mediates Antibody-Dependent Cellular Cytotoxicity. Cell Host Microbe 2019, 25, 578–587.e5. [Google Scholar] [CrossRef]
- Pancera, M.; Wyatt, R. Selective recognition of oligomeric HIV-1 primary isolate envelope glycoproteins by potently neutralizing ligands requires efficient precursor cleavage. Virology 2005, 332, 145–156. [Google Scholar] [CrossRef]
- Ledgerwood, J.E.; Coates, E.E.; Yamshchikov, G.; Saunders, J.G.; Holman, L.; Enama, M.E.; DeZure, A.; Lynch, R.M.; Gordon, I.; Plummer, S.; et al. Safety, pharmacokinetics and neutralization of the broadly neutralizing HIV-1 human monoclonal antibody VRC01 in healthy adults. Clin. Exp. Immunol. 2015, 182, 289–301. [Google Scholar] [PubMed]
- Schoofs, T.; Klein, F.; Braunschweig, M.; Kreider, E.F.; Feldmann, A.; Nogueira, L.; Oliveira, T.; Lorenzi, J.C.; Parrish, E.H.; Learn, G.H.; et al. HIV-1 therapy with monoclonal antibody 3BNC117 elicits host immune responses against HIV-1. Science 2016, 352, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Cahn, P.; Fink, V.; Patterson, P. Fostemsavir: A new CD4 attachment inhibitor. Curr. Opin. HIV AIDS 2018, 13, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Muccini, C.; Canetti, D.; Castagna, A.; Spagnuolo, V. Efficacy and Safety Profile of Fostemsavir for the Treatment of People with Human Immunodeficiency Virus-1 (HIV-1): Current Evidence and Place in Therapy. Drug Des. Dev. Ther. 2022, 16, 297–304. [Google Scholar] [CrossRef]
- Pancera, M.; Lai, Y.T.; Bylund, T.; Druz, A.; Narpala, S.; O’Dell, S.; Schön, A.; Bailer, R.T.; Chuang, G.Y.; Geng, H.; et al. Crystal structures of trimeric HIV envelope with entry inhibitors BMS-378806 and BMS-626529. Nat. Chem. Biol. 2017, 13, 1115–1122. [Google Scholar] [CrossRef]
- Prévost, J.; Chen, Y.; Zhou, F.; Tolbert, W.D.; Gasser, R.; Medjahed, H.; Gottumukkala, S.; Hessell, A.J.; Rao, V.B.; Pozharski, E.; et al. Structure-function Analyses Reveal Key Molecular Determinants of HIV-1 CRF01_AE Resistance to the Entry Inhibitor Temsavir. bioRxiv 2023. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, K.; Wang, W.L.; Nguyen, H.T.; Chen, S.; Lu, M.; Go, E.P.; Ding, H.; Steinbock, R.T.; Desaire, H.; et al. Asymmetric structures and conformational plasticity of the uncleaved full-length human immunodeficiency virus (HIV-1) envelope glycoprotein trimer. J. Virol. 2021, 95, e0052921. [Google Scholar] [CrossRef]
- Boutin, M.; Vézina, D.; Ding, S.; Prévost, J.; Laumaea, A.; Marchitto, L.; Anand, S.P.; Medjahed, H.; Gendron-Lepage, G.; Bourassa, C.; et al. Temsavir Treatment of HIV-1-Infected Cells Decreases Envelope Glycoprotein Recognition by Broadly Neutralizing Antibodies. mBio 2022, 13, e0057722. [Google Scholar] [CrossRef]
- Veillette, M.; Désormeaux, A.; Medjahed, H.; Gharsallah, N.-E.; Coutu, M.; Baalwa, J.; Guan, Y.; Lewis, G.; Ferrari, G.; Hahn, B.H.; et al. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J. Virol. 2014, 88, 2633–2644. [Google Scholar] [CrossRef]
- Emi, N.; Friedmann, T.; Yee, J.K. Pseudotype formation of murine leukemia virus with the G protein of vesicular stomatitis virus. J. Virol. 1991, 65, 1202–1207. [Google Scholar] [CrossRef]
- Mao, Y.; Wang, L.; Gu, C.; Herschhorn, A.; Xiang, S.-H.; Haim, H.; Yang, X.; Sodroski, J. Subunit organization of the membrane-bound HIV-1 envelope glycoprotein trimer. Nat. Struct. Mol. Biol. 2012, 19, 893–899. [Google Scholar] [CrossRef]
- Wu, X.; Parast, A.B.; Richardson, B.A.; Nduati, R.; John-Stewart, G.; Mbori-Ngacha, D.; Rainwater, S.M.; Overbaugh, J. Neutralization escape variants of human immunodeficiency virus type 1 are transmitted from mother to infant. J. Virol. 2006, 80, 835–844. [Google Scholar] [CrossRef]
- Finzi, A.; Xiang, S.-H.; Pacheco, B.; Wang, L.; Haight, J.; Kassa, A.; Danek, B.; Pancera, M.; Kwong, P.D.; Sodroski, J. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol. Cell 2010, 37, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Keele, B.F.; Learn, G.H.; Giorgi, E.E.; Li, H.; Decker, J.M.; Wang, S.; Baalwa, J.; Kraus, M.H.; et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009, 206, 1273–1289. [Google Scholar] [CrossRef] [PubMed]
- Ochsenbauer, C.; Edmonds, T.G.; Ding, H.; Keele, B.F.; Decker, J.; Salazar, M.G.; Salazar-Gonzalez, J.F.; Shattock, R.; Haynes, B.F.; Shaw, G.M.; et al. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J. Virol. 2012, 86, 2715–2728. [Google Scholar] [CrossRef] [PubMed]
- Parrish, N.F.; Wilen, C.B.; Banks, L.B.; Iyer, S.S.; Pfaff, J.M.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Decker, J.M.; Parrish, E.H.; Berg, A.; et al. Transmitted/founder and chronic subtype C HIV-1 use CD4 and CCR5 receptors with equal efficiency and are not inhibited by blocking the integrin α4β7. PLoS Pathog. 2012, 8, e1002686. [Google Scholar] [CrossRef]
- Parrish, N.F.; Gao, F.; Li, H.; Giorgi, E.E.; Barbian, H.J.; Parrish, E.H.; Zajic, L.; Iyer, S.S.; Decker, J.M.; Kumar, A. Phenotypic properties of transmitted founder HIV-1. Proc. Natl. Acad. Sci. USA 2013, 110, 6626–6633. [Google Scholar] [CrossRef]
- O’Brien, W.A.; Koyanagi, Y.; Namazie, A.; Zhao, J.Q.; Diagne, A.; Idler, K.; Zack, J.A.; Chen, I.S. HIV-1 tropism for mononuclear phagocytes can be determined by regions of gp120 outside the CD4-binding domain. Nature 1990, 348, 69–73. [Google Scholar] [CrossRef]
- Li, Y.; Kappes, J.C.; Conway, J.A.; Price, R.W.; Shaw, G.; Hahn, B. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: Identification of replication-competent and-defective viral genomes. J. Virol. 1991, 65, 3973–3985. [Google Scholar] [CrossRef]
- Theodore, T.S.; Englund, G.; Buckler-White, A.; Buckler, C.E.; Martin, M.A.; Peden, K.W. Construction and characterization of a stable full-length macrophage-tropic HIV type 1 molecular clone that directs the production of high titers of progeny virions. AIDS Res. Hum. Retrovir. 1996, 12, 191–194. [Google Scholar] [CrossRef]
- Sanders, R.W.; Derking, R.; Cupo, A.; Julien, J.-P.; Yasmeen, A.; de Val, N.; Kim, H.J.; Blattner, C.; de la Peña, A.T.; Korzun, J.; et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP. 664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 2013, 9, e1003618. [Google Scholar] [CrossRef] [PubMed]
- Prévost, J.; Zoubchenok, D.; Richard, J.; Veillette, M.; Pacheco, B.; Coutu, M.; Brassard, N.; Parsons, M.S.; Ruxrungtham, K.; Bunupuradah, T.; et al. Influence of the envelope gp120 Phe 43 cavity on HIV-1 sensitivity to antibody-dependent cell-mediated cytotoxicity responses. J. Virol. 2017, 91, e02452-16. [Google Scholar] [CrossRef] [PubMed]
- Prévost, J.; Medjahed, H.; Vézina, D.; Chen, H.-C.; Hahn, B.H.; Smith, A.B.; Finzi, A. HIV-1 Envelope Glycoproteins Proteolytic Cleavage Protects Infected Cells from ADCC Mediated by Plasma from Infected Individuals. Viruses 2021, 13, 2236. [Google Scholar] [CrossRef]
- Crooks, E.T.; Tong, T.; Osawa, K.; Binley, J.M. Enzyme digests eliminate nonfunctional Env from HIV-1 particle surfaces, leaving native Env trimers intact and viral infectivity unaffected. J. Virol. 2011, 85, 5825–5839. [Google Scholar] [CrossRef]
- Haim, H.; Salas, I.; Sodroski, J. Proteolytic processing of the human immunodeficiency virus envelope glycoprotein precursor decreases conformational flexibility. J. Virol. 2013, 87, 1884–1889. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Menendez, L.R.; Witt, K.; Espy, N.; Princiotto, A.; Madani, N.; Pacheco, B.; Finzi, A.; Sodroski, J. Comparison of uncleaved and mature human immunodeficiency virus membrane envelope glycoprotein trimers. J. Virol. 2018, 92, e00277-18. [Google Scholar] [CrossRef]
- Ringe, R.P.; Sanders, R.W.; Yasmeen, A.; Kim, H.J.; Lee, J.H.; Cupo, A.; Korzun, J.; Derking, R.; van Montfort, T.; Julien, J.-P.; et al. Cleavage strongly influences whether soluble HIV-1 envelope glycoprotein trimers adopt a native-like conformation. Proc. Natl. Acad. Sci. USA 2013, 110, 18256–18261. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, B.K.; Pancera, M.; Phogat, S.; O’Dell, S.; McKee, K.; Guenaga, J.; Robinson, J.; Mascola, J.; Wyatt, R.T. HIV type 1 Env precursor cleavage state affects recognition by both neutralizing and nonneutralizing gp41 antibodies. AIDS Res. Hum. Retrovir. 2011, 27, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Ma, X.; Reichard, N.; Terry, D.S.; Arthos, J.; Smith, A.B., III; Sodroski, J.G.; Blanchard, S.C.; Mothes, W. Shedding-resistant HIV-1 envelope glycoproteins adopt downstream conformations that remain responsive to conformation-preferring ligands. J. Virol. 2020, 94, e00597-20. [Google Scholar] [CrossRef]
- Madani, N.; Perdigoto, A.L.; Srinivasan, K.; Cox, J.M.; Chruma, J.J.; LaLonde, J.; Head, M.; Smith, A.B., III; Sodroski, J.G. Localized changes in the gp120 envelope glycoprotein confer resistance to human immunodeficiency virus entry inhibitors BMS-806 and #155. J. Virol. 2004, 78, 3742–3752. [Google Scholar] [CrossRef]
- Richard, J.; Prévost, J.; Bourassa, C.; Brassard, N.; Boutin, M.; Benlarbi, M.; Goyette, G.; Medjahed, H.; Gendron-Lepage, G.; Gaudette, F.; et al. Temsavir blocks the immunomodulatory activities of HIV-1 soluble gp120. Cell Chem. Biol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.; Prévost, J.; Alsahafi, N.; Ding, S.; Finzi, A. Impact of HIV-1 envelope conformation on ADCC responses. Trends Microbiol. 2018, 26, 253–265. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boutin, M.; Medjahed, H.; Nayrac, M.; Lotke, R.; Gendron-Lepage, G.; Bourassa, C.; Sauter, D.; Richard, J.; Finzi, A. Temsavir Modulates HIV-1 Envelope Conformation by Decreasing Its Proteolytic Cleavage. Viruses 2023, 15, 1189. https://doi.org/10.3390/v15051189
Boutin M, Medjahed H, Nayrac M, Lotke R, Gendron-Lepage G, Bourassa C, Sauter D, Richard J, Finzi A. Temsavir Modulates HIV-1 Envelope Conformation by Decreasing Its Proteolytic Cleavage. Viruses. 2023; 15(5):1189. https://doi.org/10.3390/v15051189
Chicago/Turabian StyleBoutin, Marianne, Halima Medjahed, Manon Nayrac, Rishikesh Lotke, Gabrielle Gendron-Lepage, Catherine Bourassa, Daniel Sauter, Jonathan Richard, and Andrés Finzi. 2023. "Temsavir Modulates HIV-1 Envelope Conformation by Decreasing Its Proteolytic Cleavage" Viruses 15, no. 5: 1189. https://doi.org/10.3390/v15051189