
Tracking the Genomic Evolution of SARS-CoV-2 for 29 Months in South Korea

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Sample Collection
2.2. Sample Processing and Sequencing
2.3. Sequence and Statistical Analysis
3. Results
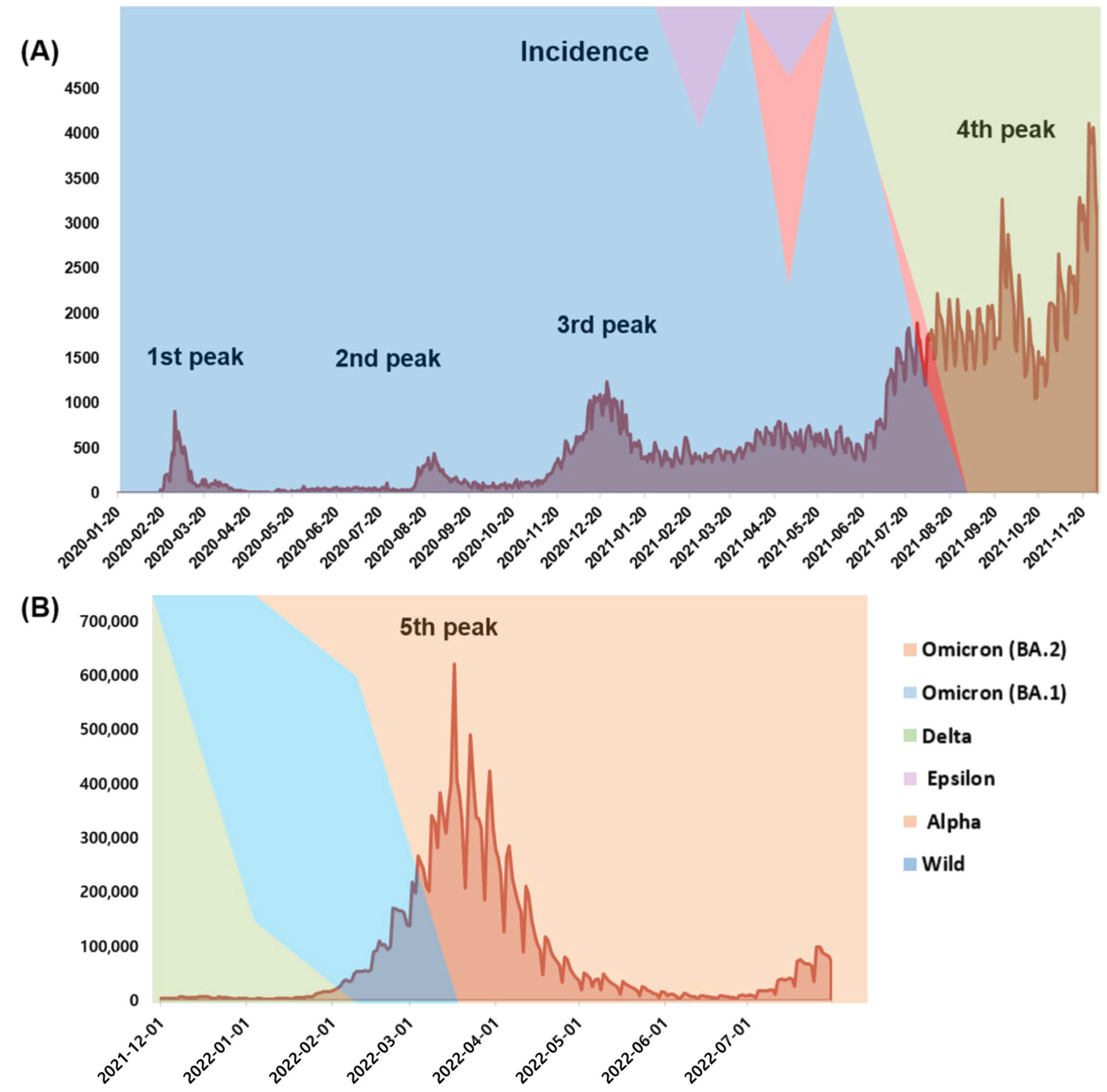
3.1. Incidence and Mortality during the COVID-19 Pandemic
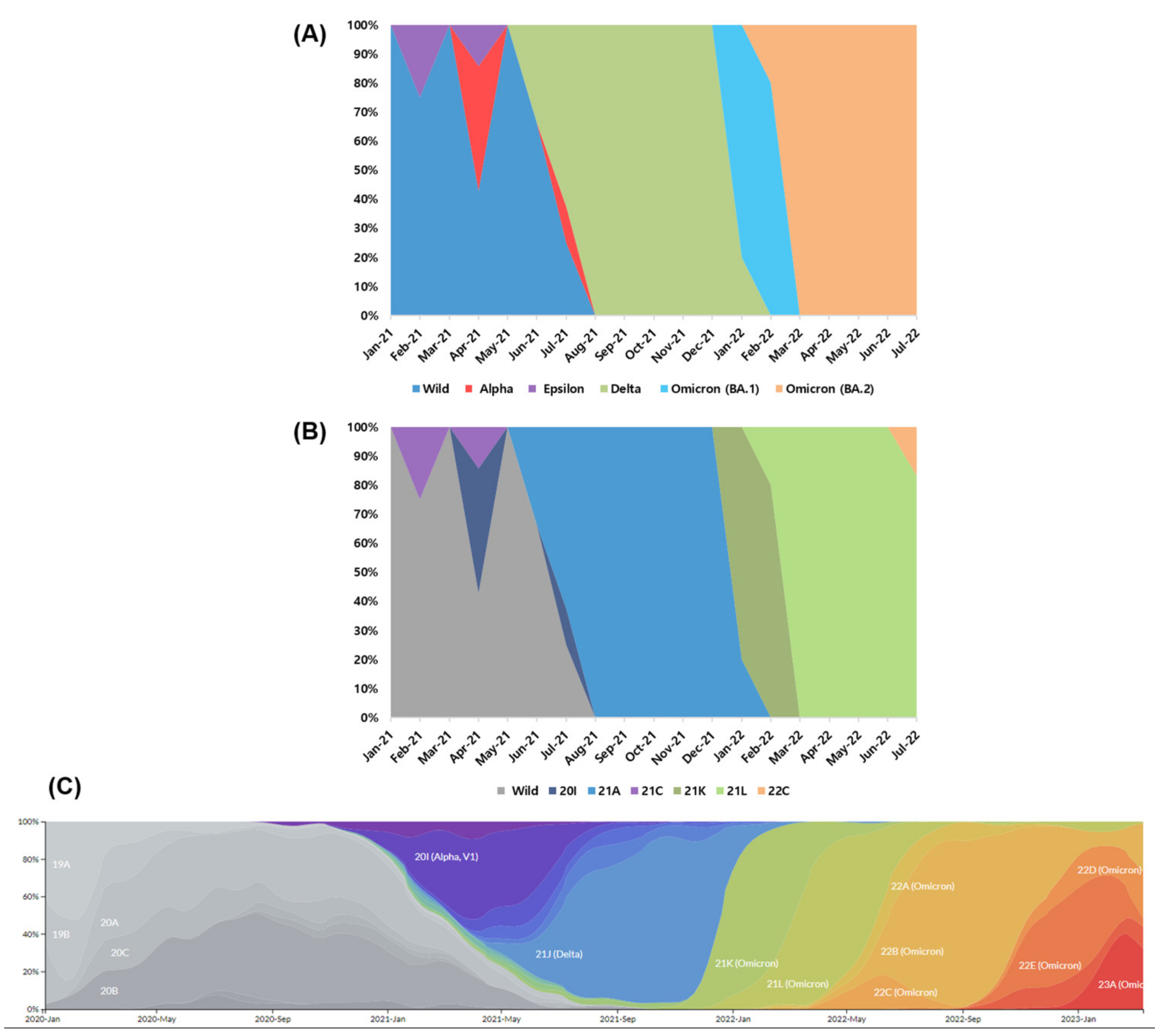
3.2. Strain Classification
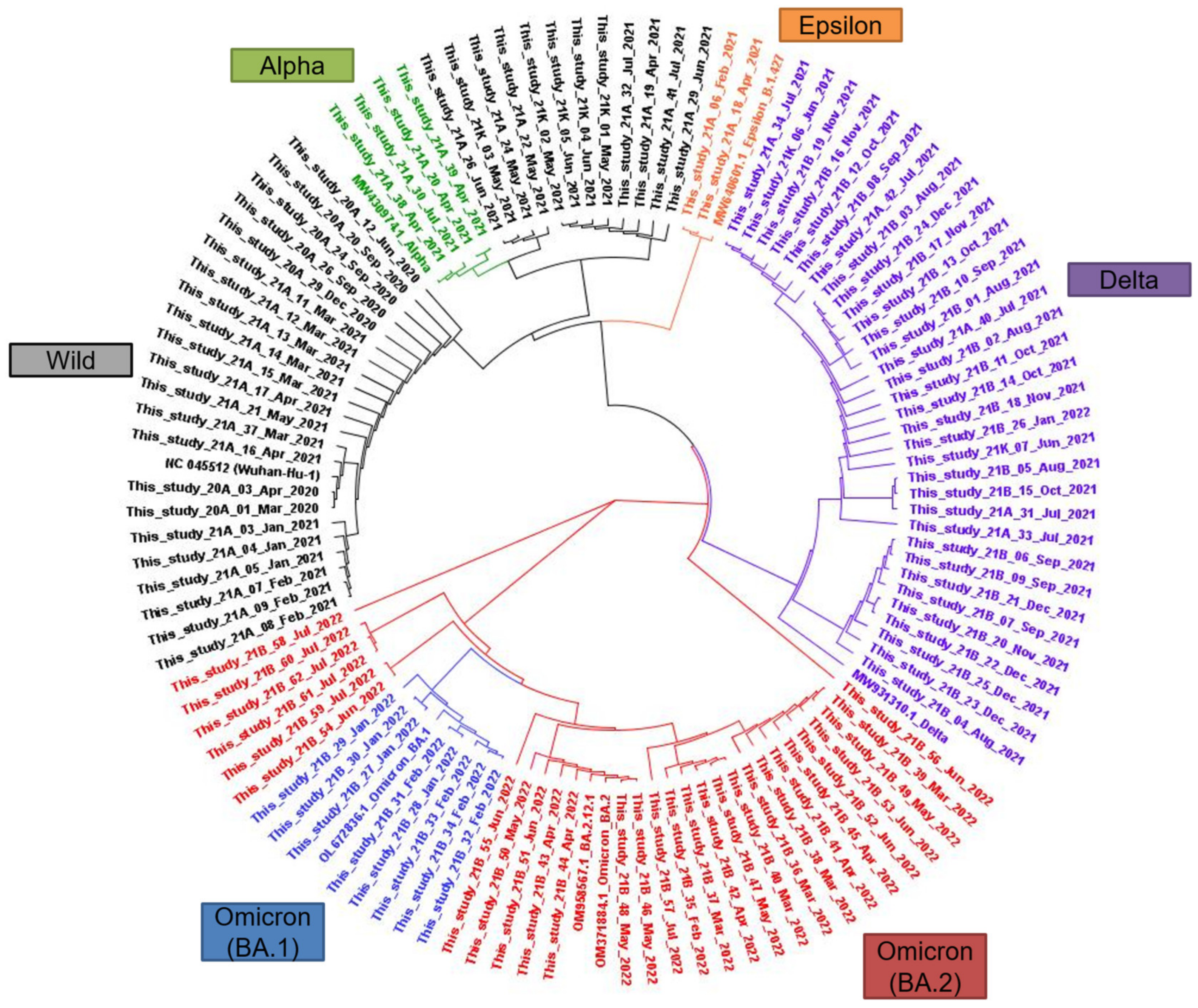
3.3. Phylogenetic Relationships
3.4. Major Mutations Related to Variants of Concern
3.5. Uncommon Mutations According to the WHO Classification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hirotsu, Y.; Omata, M. SARS-CoV-2 B.1.1.7 lineage rapidly spreads and replaces R.1 lineage in Japan: Serial and stationary observation in a community. Infect. Genet. Evol. 2021, 95, 105088. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Omata, M. Discovery of a SARS-CoV-2 variant from the P.1 lineage harboring K417T/E484K/N501Y mutations in Kofu, Japan. J. Infect. 2021, 82, 276–316. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Coronnavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 1 September 2022).
- KCDC. KCDC Coronnavirus (COVID-19) Dashboard. Available online: http://ncov.mohw.go.kr/ (accessed on 1 September 2022).
- WHO. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 11 August 2022).
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, A.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Singh, J.; Malhotra, A.G.; Biswas, D.; Shankar, P.; Lokhande, L.; Yadav, A.K.; Raghuvanshi, A.; Kale, D.; Nema, S.; Saigal, S.; et al. Relative Consolidation of the Kappa Variant Pre-Dates the Massive Second Wave of COVID-19 in India. Genes 2021, 12, 1803. [Google Scholar] [CrossRef]
- Dougherty, K.; Mannell, M.; Naqvi, O.; Matson, D.; Stone, J. SARS-CoV-2 B.1.617.2 (Delta) Variant COVID-19 Outbreak Associated with a Gymnastics Facility—Oklahoma, April–May 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 1004–1007. [Google Scholar] [CrossRef]
- Sheikh, A.; McMenamin, J.; Taylor, B.; Robertson, C. SARS-CoV-2 Delta VOC in Scotland: Demographics, risk of hospital admission, and vaccine effectiveness. Lancet 2021, 397, 2461–2462. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Islam, F.; Dhawan, M.; Nafady, M.H.; Emran, T.B.; Mitra, S.; Choudhary, O.P.; Akter, A. Understanding the omicron variant (B.1.1.529) of SARS-CoV-2: Mutational impacts, concerns, and the possible solutions. Ann. Med. Surg. (Lond.) 2022, 78, 103737. [Google Scholar] [CrossRef]
- CDC. COVID Data Tracker: Variant Proportions. Available online: https://covid.cdc.gov/covid-data-tracker/#variant-proportions (accessed on 17 September 2022).
- Khandia, R.; Singhal, S.; Alqahtani, T.; Kamal, M.A.; El-Shall, N.A.; Nainu, F.; Desingu, P.A.; Dhama, K. Emergence of SARS-CoV-2 Omicron (B.1.1.529) variant, salient features, high global health concerns and strategies to counter it amid ongoing COVID-19 pandemic. Environ. Res. 2022, 209, 112816. [Google Scholar] [CrossRef] [PubMed]
- Hachmann, N.P.; Miller, J.; Collier, A.Y.; Ventura, J.D.; Yu, J.; Rowe, M.; Bondzie, E.A.; Powers, O.; Surve, N.; Hall, K.; et al. Neutralization Escape by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4, and BA.5. N. Engl. J. Med. 2022, 387, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Rossler, A.; Riepler, L.; Bante, D.; von Laer, D.; Kimpel, J. SARS-CoV-2 Omicron Variant Neutralization in Serum from Vaccinated and Convalescent Persons. N. Engl. J. Med. 2022, 386, 698–700. [Google Scholar] [CrossRef] [PubMed]
- Takashita, E.; Yamayoshi, S.; Simon, V.; van Bakel, H.; Sordillo, E.M.; Pekosz, A.; Fukushi, S.; Suzuki, T.; Maeda, K.; Halfmann, P.; et al. Efficacy of Antibodies and Antiviral Drugs against Omicron BA.2.12.1, BA.4, and BA.5 Subvariants. N. Engl. J. Med. 2022, 387, 468–470. [Google Scholar] [CrossRef]
- Hong, K.H.; Lee, S.W.; Kim, T.S.; Huh, H.J.; Lee, J.; Kim, S.Y.; Park, J.S.; Kim, G.J.; Sung, H.; Roh, K.H.; et al. Guidelines for Laboratory Diagnosis of Coronavirus Disease 2019 (COVID-19) in Korea. Ann. Lab. Med. 2020, 40, 351–360. [Google Scholar] [CrossRef]
- Seo, J.W.; Lee, S.K.; Hong, I.H.; Choi, S.H.; Lee, J.Y.; Kim, H.S.; Kim, H.S. Molecular Epidemiology of Adenoviral Keratoconjunctivitis in Korea. Ann. Lab. Med. 2022, 42, 683–687. [Google Scholar] [CrossRef]
- Kang, D.; Choi, J.; Kim, Y.; Kwon, D. An analysis of the dynamic spatial spread of COVID-19 across South Korea. Sci. Rep. 2022, 12, 9364. [Google Scholar] [CrossRef]
- Kim, S.; Kim, M.; Lee, S.; Lee, Y.J. Discovering spatiotemporal patterns of COVID-19 pandemic in South Korea. Sci. Rep. 2021, 11, 24470. [Google Scholar] [CrossRef]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef]
- Chan, J.F.; Yuan, S.; Kok, K.H.; To, K.K.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.; Poon, R.W.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; du Plessis, L.; Liu, Z.; Hill, V.; Kang, M.; Lin, H.; Sun, J.; Francois, S.; Kraemer, M.U.G.; Faria, N.R.; et al. Genomic Epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell 2020, 181, 997–1003.e9. [Google Scholar] [CrossRef]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First Case of 2019 Novel Coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef]
- Deng, X.; Gu, W.; Federman, S.; du Plessis, L.; Pybus, O.G.; Faria, N.R.; Wang, C.; Yu, G.; Bushnell, B.; Pan, C.Y.; et al. Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science 2020, 369, 582–587. [Google Scholar] [CrossRef]
- Maurano, M.T.; Ramaswami, S.; Zappile, P.; Dimartino, D.; Boytard, L.; Ribeiro-Dos-Santos, A.M.; Vulpescu, N.A.; Westby, G.; Shen, G.; Feng, X.; et al. Sequencing identifies multiple early introductions of SARS-CoV-2 to the New York City Region. medRxiv 2020. [Google Scholar] [CrossRef]
- Jeong, S.; Lee, N.; Lee, S.K.; Cho, E.J.; Hyun, J.; Park, M.J.; Song, W.; Kim, H.S. STANDARD M10 SARS-CoV-2 Assay for Rapid Detection of SARS-CoV-2: Comparison of Four Real-Time PCR Assays. Diagnostics 2022, 12, 1998. [Google Scholar] [CrossRef]
- Jhun, H.; Park, H.Y.; Hisham, Y.; Song, C.S.; Kim, S. SARS-CoV-2 Delta (B.1.617.2) Variant: A Unique T478K Mutation in Receptor Binding Motif (RBM) of Spike Gene. Immune Netw. 2021, 21, e32. [Google Scholar] [CrossRef]
- Davies, N.G.; Jarvis, C.I.; Group, C.C.-W.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Shen, X.; Tang, H.; McDanal, C.; Wagh, K.; Fischer, W.; Theiler, J.; Yoon, H.; Li, D.; Haynes, B.F.; Sanders, K.O.; et al. SARS-CoV-2 variant B.1.1.7 is susceptible to neutralizing antibodies elicited by ancestral spike vaccines. Cell Host Microbe 2021, 29, 529–539.e3. [Google Scholar] [CrossRef]
- Ehelepola, N.D.B.; Wijewardana, B.A.S. Vaccine Breakthrough COVID-19 Outbreak in Section of a Hospital with 88% Attack Rate: Lessons to Be Learned. COVID 2023, 3, 226–237. [Google Scholar] [CrossRef]
- Twohig, K.A.; Nyberg, T.; Zaidi, A.; Thelwall, S.; Sinnathamby, M.A.; Aliabadi, S.; Seaman, S.R.; Harris, R.J.; Hope, R.; Lopez-Bernal, J.; et al. Hospital admission and emergency care attendance risk for SARS-CoV-2 delta (B.1.617.2) compared with alpha (B.1.1.7) variants of concern: A cohort study. Lancet Infect. Dis. 2022, 22, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Fisman, D.N.; Tuite, A.R. Evaluation of the relative virulence of novel SARS-CoV-2 variants: A retrospective cohort study in Ontario, Canada. CMAJ 2021, 193, E1619–E1625. [Google Scholar] [CrossRef] [PubMed]
- Fowlkes, A.; Gaglani, M.; Groover, K.; Thiese, M.S.; Tyner, H.; Ellingson, K.; Cohorts, H.-R. Effectiveness of COVID-19 Vaccines in Preventing SARS-CoV-2 Infection Among Frontline Workers Before and During B.1.617.2 (Delta) Variant Predominance—Eight U.S. Locations, December 2020–August 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- El Mazouri, S.; Bendani, H.; Boumajdi, N.; Aanniz, T.; Kandoussi, I.; Amzazi, S.; Belyamani, L.; Ibrahimi, A.; Ouadghiri, M. Phylogeography and genomic analysis of SARS-CoV-2 delta variant in Morocco. J. Infect. Dev. Ctries. 2022, 16, 1258–1268. [Google Scholar] [CrossRef]
- Mannsverk, S.; Bergholm, J.; Palanisamy, N.; Ellstrom, P.; Kaden, R.; Lindh, J.; Lennerstrand, J. SARS-CoV-2 variants of concern and spike protein mutational dynamics in a Swedish cohort during 2021, studied by Nanopore sequencing. Virol. J. 2022, 19, 164. [Google Scholar] [CrossRef]
- Umair, M.; Ikram, A.; Rehman, Z.; Haider, S.A.; Ammar, M.; Badar, N.; Ali, Q.; Rana, M.S.; Salman, M. Genomic diversity of SARS-CoV-2 in Pakistan during the fourth wave of pandemic. J. Med. Virol. 2022, 94, 4869–4877. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-Gonzalez, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation. medRxiv 2021. [Google Scholar] [CrossRef]
- Martin Webb, L.; Matzinger, S.; Grano, C.; Kawasaki, B.; Stringer, G.; Bankers, L.; Herlihy, R. Identification of and Surveillance for the SARS-CoV-2 Variants B.1.427 and B.1.429—Colorado, January-March 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 717–718. [Google Scholar] [CrossRef]
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science 2021, 373, 648–654. [Google Scholar] [CrossRef]
- Kumar, S.; Karuppanan, K.; Subramaniam, G. Omicron (BA.1) and Sub-Variants (BA.1, BA.2 and BA.3) of SARS-CoV-2 Spike Infectivity and Pathogenicity: A Comparative Sequence and Structural-based Computational Assessment. bioRxiv 2022, 94, 4780–4791. [Google Scholar] [CrossRef]
- Baker, J.M.; Nakayama, J.Y.; O’Hegarty, M.; McGowan, A.; Teran, R.A.; Bart, S.M.; Mosack, K.; Roberts, N.; Campos, B.; Paegle, A.; et al. SARS-CoV-2 B.1.1.529 (Omicron) Variant Transmission Within Households—Four U.S. Jurisdictions, November 2021–February 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 341–346. [Google Scholar] [CrossRef]
- Jorgensen, S.B.; Nygard, K.; Kacelnik, O.; Telle, K. Secondary Attack Rates for Omicron and Delta Variants of SARS-CoV-2 in Norwegian Households. JAMA 2022, 327, 1610–1611. [Google Scholar] [CrossRef]
- Cianci, R.; Franza, L.; Pignataro, G.; Massaro, M.G.; Rio, P.; Tota, A.; Ocarino, F.; Sacco Fernandez, M.; Franceschi, F.; Gasbarrini, A.; et al. Effect of COVID-19 Vaccination on the In-Hospital Prognosis of Patients Admitted during Delta and Omicron Waves in Italy. Vaccines 2023, 11, 373. [Google Scholar] [CrossRef]
- Sheward, D.J.; Kim, C.; Ehling, R.A.; Pankow, A.; Castro Dopico, X.; Dyrdak, R.; Martin, D.P.; Reddy, S.T.; Dillner, J.; Karlsson Hedestam, G.B.; et al. Neutralisation sensitivity of the SARS-CoV-2 omicron (B.1.1.529) variant: A cross-sectional study. Lancet Infect. Dis. 2022, 22, 813–820. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- Quadir, N.; Singh, J.; Alam, A.; Malik, A.A.; Rahman, S.A.; Hira, S.; Ehtesham, N.Z.; Sundar, D.; Hasnain, S.E. Evolution of SARS-CoV-2: BA.4/BA.5 Variants Continues to Pose New Challenges. Viruses 2022, 14, 2610. [Google Scholar] [CrossRef]
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliuc, A.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M.E.; et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 2022, 185, 2422–2433.e13. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | Wild | Alpha | Delta | Epsilon | Omicron (BA.1) | Omicron (BA.2) | Omicron (BA.2) |
|---|---|---|---|---|---|---|---|
| 21I | 21A | 21C | 21K | 21L | 22C | ||
| B.1.1.7 | B.1.617.2 | B.1.427/B.1.429 | BA.1.1 | BA.2 | BA.2.12.1 | ||
| 2020.01–2020.12 | 100% (7/7) | 0% (0/7) | 0% (0/7) | 0% (0/7) | 0% (0/7) | 0% (0/7) | 0% (0/7) |
| 2021.01–2021.06 | 78.1% (25/32) | 9.4% (3/32) | 6.3% (2/32) | 6.3% (2/32) | 0% (0/32) | 0% (0/32) | 0% (0/32) |
| 2021.07–2021.12 | 6.1% (2/33) | 3.0% (1/33) | 90.9% (30/33) | 0% (0/33) | 0% (0/33) | 0% (0/33) | 0% (0/33) |
| 2022.01–2022.07 | 0% (0/37) | 0% (0/37) | 2.7% (1/37) | 0% (0/37) | 21.6% (8/37) | 73.0% (27/37) | 2.7% (1/37) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, S.; Kim, J.-S.; Lee, S.K.; Cho, E.-J.; Hyun, J.; Song, W.; Kim, H.S. Tracking the Genomic Evolution of SARS-CoV-2 for 29 Months in South Korea. Viruses 2023, 15, 873. https://doi.org/10.3390/v15040873
Jeong S, Kim J-S, Lee SK, Cho E-J, Hyun J, Song W, Kim HS. Tracking the Genomic Evolution of SARS-CoV-2 for 29 Months in South Korea. Viruses. 2023; 15(4):873. https://doi.org/10.3390/v15040873
Chicago/Turabian StyleJeong, Seri, Jae-Seok Kim, Su Kyung Lee, Eun-Jung Cho, Jungwon Hyun, Wonkeun Song, and Hyun Soo Kim. 2023. "Tracking the Genomic Evolution of SARS-CoV-2 for 29 Months in South Korea" Viruses 15, no. 4: 873. https://doi.org/10.3390/v15040873