
Cross-Hemispheric Genetic Diversity and Spatial Genetic Structure of Callinectes sapidus Reovirus 1 (CsRV1)
 , , ,
, , ,
Abstract
:1. Introduction
1.1. Marine Disease Transmission
1.2. Blue Crab–CsRV1 Pathosystem
2. Materials and Methods
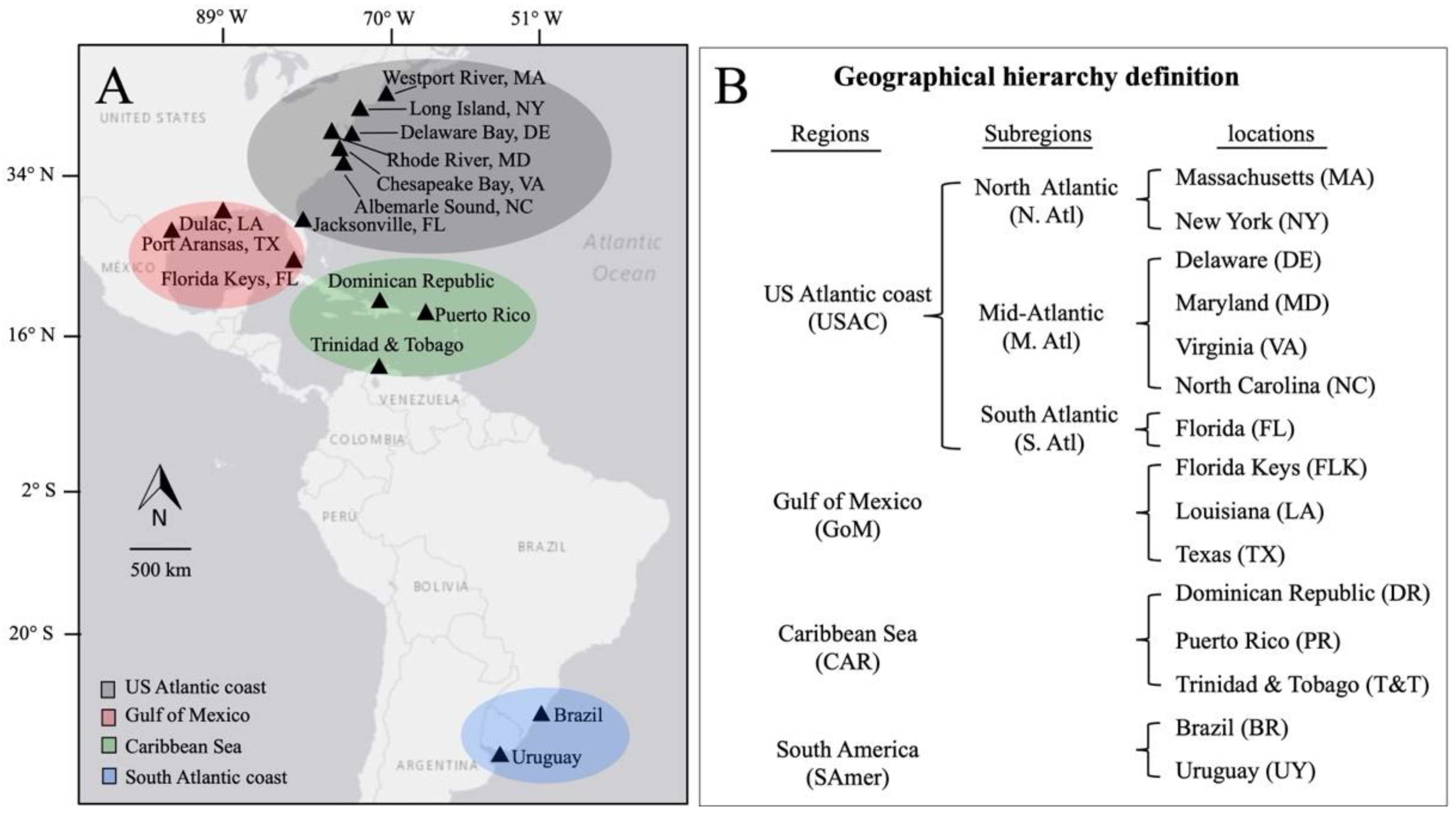
2.1. Sampling
2.2. Sequencing
2.3. Sequence Editing and Alignment
2.4. Phylogenetic Analyses
2.5. Spatial Patterns of Genetic Variability and Population Structure
2.6. Analyses of Nucleotide Diversity
2.7. Neutrality Tests and Population Differentiation
3. Results
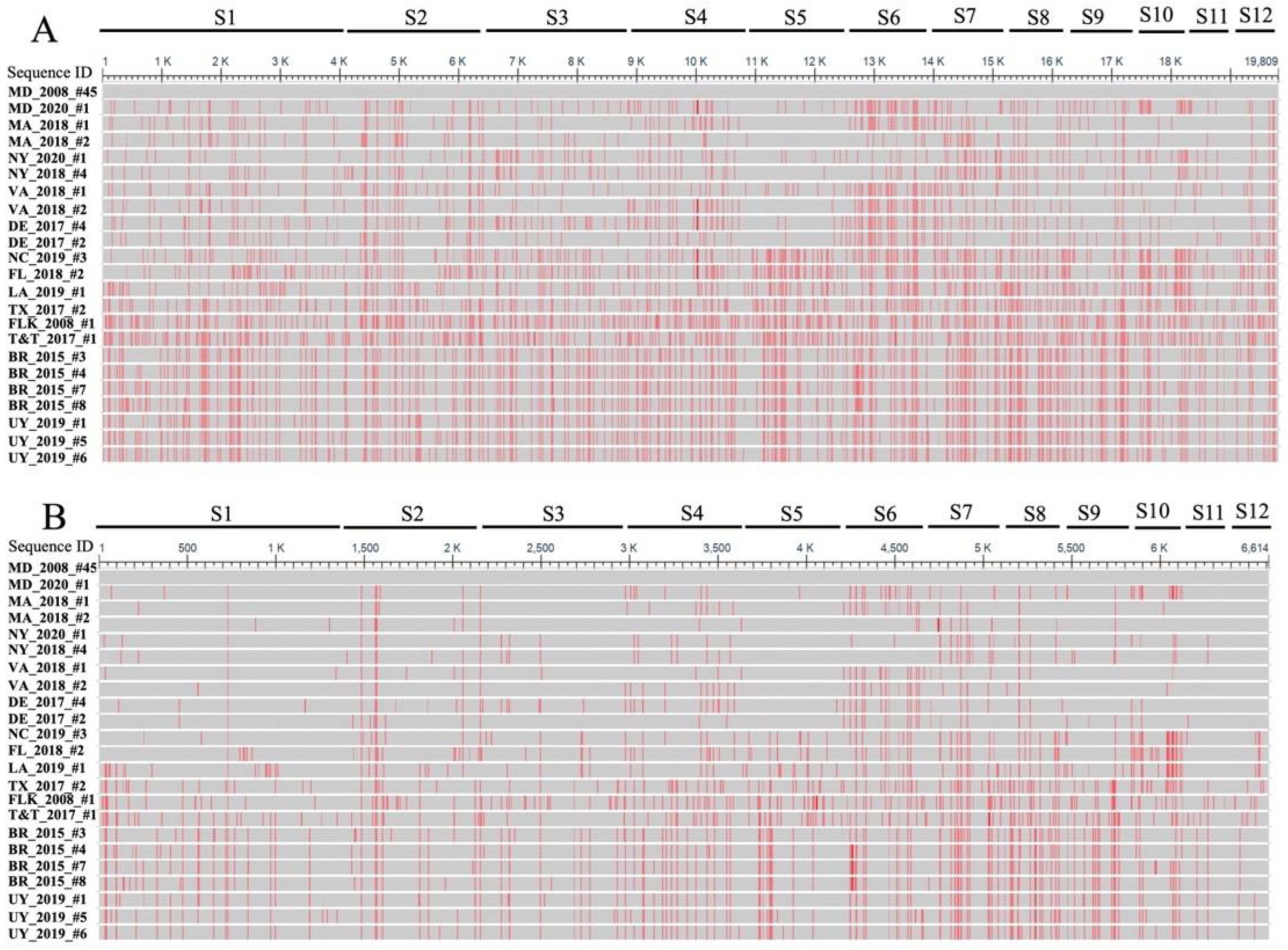
3.1. CsRV1 Genome Sequences
3.2. CsRV1 Phylogenetics
3.3. Genetic Diversity and Selection
3.4. Neutrality Tests and Genetic Differentiation
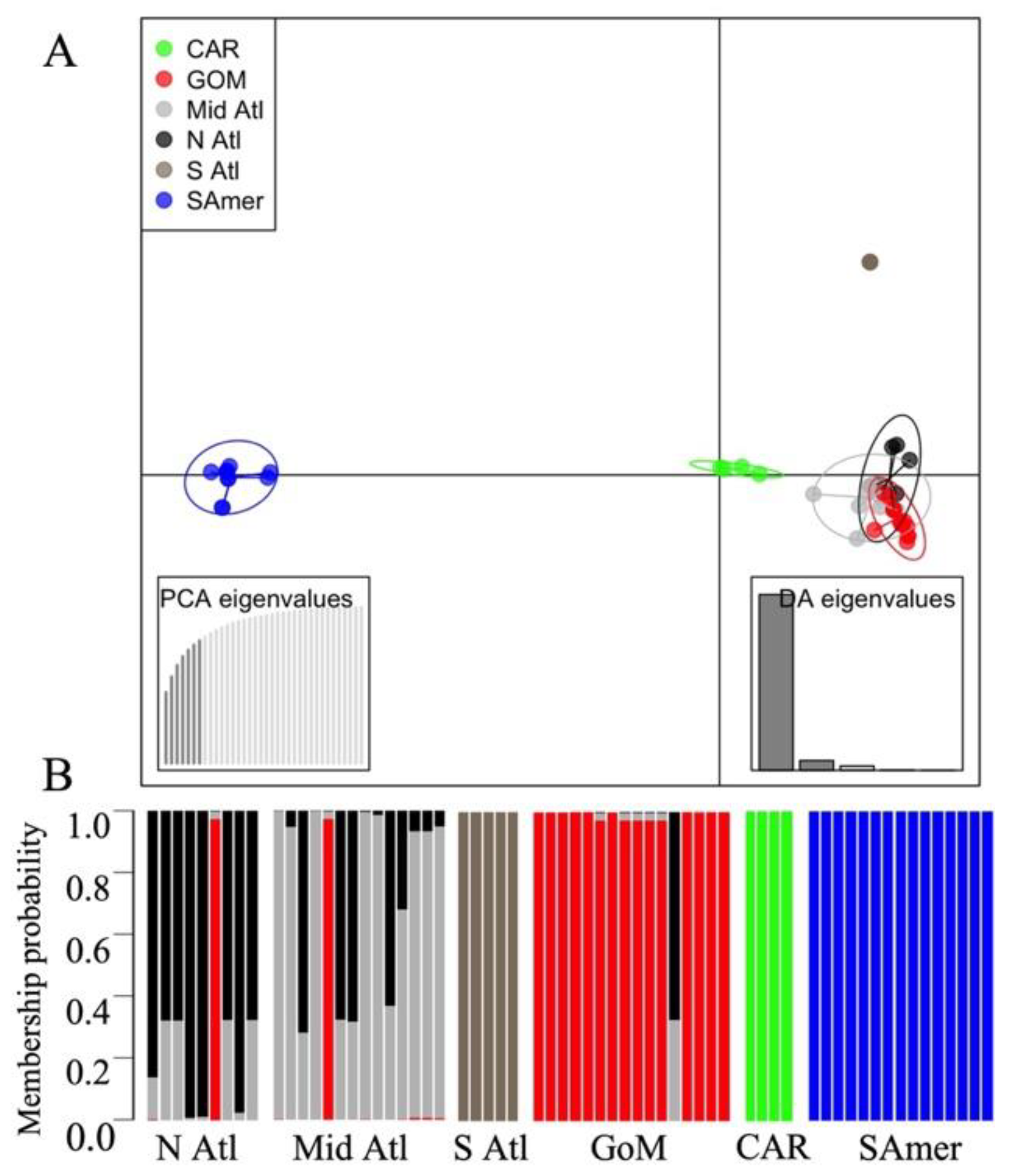
3.5. Spatial Genetic Structure and Admixture Patterns
4. Discussion
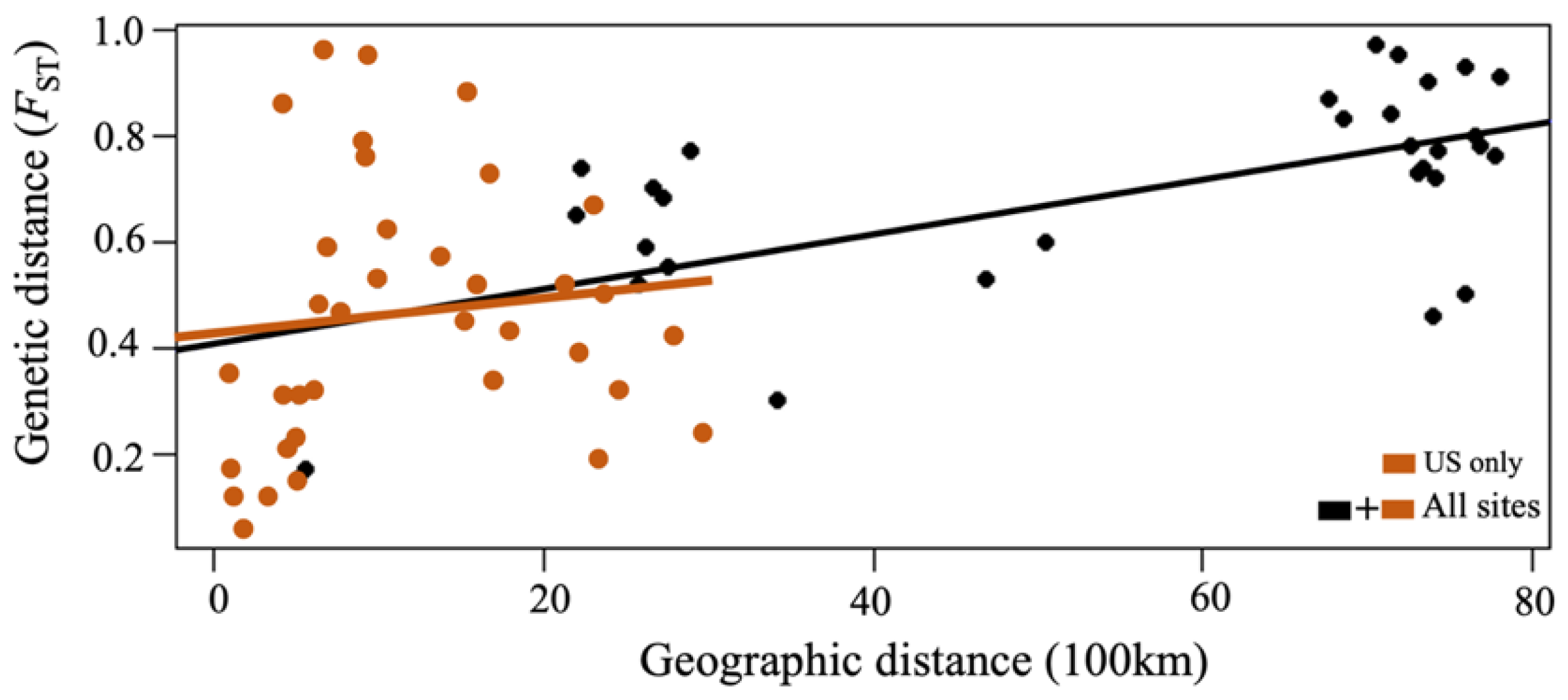
4.1. Genetic Differentiation between CsRV1 Populations over Large Geographic Scales
4.2. Limited Genetic Differentiation Found over Small Geographic Scales
4.3. Anthropogenic Transport of CsRV1?
4.4. Genetic Diversity, Variable Environment and Host Life History
4.5. Variable Evolution among CsRV1 Genomic Segments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lafferty, K.D.; Porter, J.W.; Ford, S.E. Are Diseases Increasing in the Ocean? Annu. Rev. Ecol. Evol. Syst. 2004, 35, 31–54. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Behringer, D.C.; Bojko, J.; Kough, A.S.; Plough, L.; dos Santos Tavares, C.P.; Aguilar-Perera, A.; Reynoso, O.S.; Seepersad, G.; Maharaj, O.; et al. Climate and Season Are Associated with Prevalence and Distribution of Trans-Hemispheric Blue Crab Reovirus (Callinectes Sapidus Reovirus 1). Mar. Ecol. Prog. Ser. 2020, 647, 123–133. [Google Scholar] [CrossRef]
- Lessios, H. Mass Mortality Of Diadema-Antillarum In The Caribbean: What Have We Learned. Annu. Rev. Ecol. Syst. 1988, 19, 371–393. [Google Scholar] [CrossRef]
- Aronson, R.B.; Precht, W.F. White-Band Disease and the Changing Face of Caribbean Coral Reefs. Hydrobiologia 2001, 460, 25–38. [Google Scholar] [CrossRef]
- Muller, E.M.; Sartor, C.; Alcaraz, N.I.; van Woesik, R. Spatial Epidemiology of the Stony-Coral-Tissue-Loss Disease in Florida. Front. Mar. Sci. 2020, 7, 163. [Google Scholar] [CrossRef] [Green Version]
- Groner, M.L.; Maynard, J.; Breyta, R.; Carnegie, R.B.; Dobson, A.; Friedman, C.S.; Froelich, B.; Garren, M.; Gulland, F.M.D.; Heron, S.F.; et al. Managing Marine Disease Emergencies in an Era of Rapid Change. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150364. [Google Scholar] [CrossRef] [Green Version]
- Biek, R.; Real, L.A. The Landscape Genetics of Infectious Disease Emergence and Spread. Mol. Ecol. 2010, 19, 3515–3531. [Google Scholar] [CrossRef] [Green Version]
- Carver, S.; Lunn, T. When Are Pathogen Dynamics Likely to Reflect Host Population Genetic Structure? Mol. Ecol. 2020, 29, 859–861. [Google Scholar] [CrossRef] [Green Version]
- Olival, K.J.; Latinne, A.; Islam, A.; Epstein, J.H.; Hersch, R.; Engstrand, R.C.; Gurley, E.S.; Amato, G.; Luby, S.P.; Daszak, P. Population Genetics of Fruit Bat Reservoir Informs the Dynamics, Distribution and Diversity of Nipah Virus. Mol. Ecol. 2020, 29, 970–985. [Google Scholar] [CrossRef]
- Daversa, D.R.; Fenton, A.; Dell, A.I.; Garner, T.W.J.; Manica, A. Infections on the Move: How Transient Phases of Host Movement Influence Disease Spread. Proc. R. Soc. B Biol. Sci. 2017, 284, 20171807. [Google Scholar] [CrossRef] [Green Version]
- Mazé-Guilmo, E.; Blanchet, S.; Mccoy, K.D.; Loot, G. Host Dispersal as the Driver of Parasite Genetic Structure: A Paradigm Lost? Ecol. Lett. 2016, 19, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Fountain-Jones, N.M.; Kraberger, S.; Gagne, R.B.; Trumbo, D.R.; Salerno, P.E.; Chris Funk, W.; Crooks, K.; Biek, R.; Alldredge, M.; Logan, K.; et al. Host Relatedness and Landscape Connectivity Shape Pathogen Spread in the Puma, a Large Secretive Carnivore. Commun. Biol. 2021, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.E.T.; Suttle, C.A. Biogeography of Viruses in the Sea. Annu. Rev. Virol. 2015, 2, 41–66. [Google Scholar] [CrossRef] [PubMed]
- McCallum, H.; Harvell, D.; Dobson, A. Rates of Spread of Marine Pathogens. Ecol. Lett. 2003, 6, 1062–1067. [Google Scholar] [CrossRef]
- McCallum, H.; Kuris, A.; Harvell, C.; Lafferty, K.; Smith, G.; Porter, J. Does Terrestrial Epidemiology Apply to Marine Systems? Trends Ecol. Evol. 2004, 19, 585–591. [Google Scholar] [CrossRef]
- STRATHMANN, R.R. Why Life Histories Evolve Differently in the Sea. Am. Zool. 1990, 30, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Behringer, D.C.; Karvonen, A.; Bojko, J. Parasite Avoidance Behaviours in Aquatic Environments. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170202. [Google Scholar] [CrossRef] [Green Version]
- Gaines, S.; Brown, S.; Roughgarden, J. Spatial Variation in Larval Concentrations as a Cause of Spatial Variation in Settlement for the Barnacle, Balanus glandula. Oecologia 1985, 67, 267–272. [Google Scholar] [CrossRef]
- Roughgarden, J.; Iwasa, Y.; Baxter, C. Demographic Theory for an Open Marine Population with Space-Limited Recruitment. Ecology 1985, 66, 54–67. [Google Scholar] [CrossRef]
- Hyder, K.; Åberg, P.; Johnson, M.P.; Hawkins, S.J. Models of Open Populations with Space-Limited Recruitment: Extension of Theory and Application to the Barnacle Chthamalus montagui. J. Anim. Ecol. 2001, 70, 853–863. [Google Scholar] [CrossRef]
- van der Molen, J.; García-García, L.M.; Whomersley, P.; Callaway, A.; Posen, P.E.; Hyder, K. Connectivity of Larval Stages of Sedentary Marine Communities between Hard Substrates and Offshore Structures in the North Sea. Sci. Rep. 2018, 8, 14772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, L.; Carvalho, G.R. Paradigm Shifts in Marine Fisheries Genetics: Ugly Hypotheses Slain by Beautiful Facts. Fish Fish. 2008, 9, 333–362. [Google Scholar] [CrossRef]
- Eldon, B.; Riquet, F.; Yearsley, J.; Jollivet, D.; Broquet, T. Current Hypotheses to Explain Genetic Chaos under the Sea. Curr. Zool 2016, 62, 551–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moya, A.; Holmes, E.C.; González-Candelas, F. The Population Genetics and Evolutionary Epidemiology of RNA Viruses. Nat. Rev. Microbiol. 2004, 2, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Ewart, J.W.; Ford, S.E. History and Impact of MSX and Dermo Diseases on Oyster Stocks in the Northeast Region; Northeastern Regional Aquaculture Center, University of Massachusetts Dartmouth: Dartmouth, MA, USA, 1993. [Google Scholar]
- Andrews, J.D. Oyster Diseases in Chesapeake Bay. Mar. Fish. Rev. 1979, 41, 45–53. [Google Scholar]
- Burge, C.; Strenge, R.; Friedman, C. Detection of the Oyster Herpesvirus in Commercial Bivalves in Northern California, USA: Conventional and Quantitative PCR. Dis. Aquat. Organ. 2011, 94, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Mordecai, G.J.; Miller, K.M.; Bass, A.L.; Bateman, A.W.; Teffer, A.K.; Caleta, J.M.; di Cicco, E.; Schulze, A.D.; Kaukinen, K.H.; Li, S.; et al. Aquaculture Mediates Global Transmission of a Viral Pathogen to Wild Salmon. Sci. Adv. 2021, 7, eabe2592. [Google Scholar] [CrossRef]
- Bauer, R.T. Testing Generalizations about Latitudinal Variation in Reproduction and Recruitment Patterns with Sicyoniid and Caridean Shrimp Species. Invertebr. Reprod. Dev 1992, 22, 193–202. [Google Scholar] [CrossRef]
- Barrett, L.G.; Thrall, P.H.; Burdon, J.J.; Linde, C.C. Life History Determines Genetic Structure and Evolutionary Potential of Host–Parasite Interactions. Trends Ecol. Evol. 2008, 23, 678–685. [Google Scholar] [CrossRef] [Green Version]
- Restif, O.; Koella, J.C. Shared Control of Epidemiological Traits in a Coevolutionary Model of Host-Parasite Interactions. Am. Nat. 2003, 161, 827–836. [Google Scholar] [CrossRef]
- Gandon, S.; Buckling, A.; Decaestecker, E.; Day, T. Host-Parasite Coevolution and Patterns of Adaptation across Time and Space. J. Evol. Biol. 2008, 21, 1861–1866. [Google Scholar] [CrossRef] [Green Version]
- Obbard, D.J.; Dudas, G. The Genetics of Host–Virus Coevolution in Invertebrates. Curr. Opin. Virol. 2014, 8, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Hungria, D.B.; dos Santos Tavares, C.P.; Pereira, L.Â.; de Assis Teixeira da Silva, U.; Ostrensky, A. Global Status of Production and Commercialization of Soft-Shell Crabs. Aquac. Int. 2017, 25, 2213–2226. [Google Scholar] [CrossRef]
- NOAA. NOAA Landings. Available online: https://foss.nmfs.noaa.gov (accessed on 1 February 2023).
- Mancinelli, G.; Bardelli, R.; Zenetos, A. A Global Occurrence Database of the Atlantic Blue Crab Callinectes Sapidus. Sci. Data 2021, 8, 111. [Google Scholar] [CrossRef]
- Hines, A.H. Ecology of Juvenile and Adult Blue Crabs. In The Blue Crab: Callinectes Sapidus; Kennedy, V.S., Cronin, L.E., Eds.; Maryland Sea Grant College: College Park, MD, USA, 2007; pp. 565–654. [Google Scholar]
- Brylawski, B.J.; Miller, T.J. Temperature-Dependent Growth of the Blue Crab (Callinectes Sapidus): A Molt Process Approach. Can. J. Fish. Aquat. Sci. 2006, 63, 1298–1308. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.A.; Ortega, I.; D’Incao, F. The Importance of Shallow Areas as Nursery Grounds for the Recruitment of Blue Crab (Callinectes sapidus) Juveniles in Subtropical Estuaries of Southern Brazil. Reg. Stud. Mar. Sci. 2019, 25, 100492. [Google Scholar] [CrossRef]
- Epifanio, C.E.; Valenti, C.C.; Pembroke, A.E. Dispersal and Recruitment of Blue Crab Larvae in Delaware Bay, U.S.A. Estuar. Coast. Shelf. Sci. 1984, 18, 1–12. [Google Scholar] [CrossRef]
- Epifanio, C.E.; Garvine, R.W. Larval Transport on the Atlantic Continental Shelf of North America: A Review. Estuar. Coast. Shelf Sci. 2001, 52, 51–77. [Google Scholar] [CrossRef]
- Oesterling, M.J.; Adams, C.A. Migration of Blue Crabs along Florida’s Gulf Coast. Blue Crab Colloquium; Perry, H.M., van Engel, W.A., Eds.; Gulf States Marine Fisheries Commission: Biloxi, MS, USA, 16 October 1982; pp. 37–57. [Google Scholar]
- Gelpi, C.; Fry, B.; Condrey, R.; Fleeger, J.; Dubois, S. Using Δ13C and Δ15N to Determine the Migratory History of Offshore Louisiana Blue Crab Spawning Stocks. Mar. Ecol. Prog. Ser. 2013, 494, 205–218. [Google Scholar] [CrossRef] [Green Version]
- McMillen-Jackson, A.L.; Bert, T.M.; Steele, P. Population Genetics of the Blue Crab Callinectes Sapidus: Modest Population Structuring in a Background of High Gene Flow. Mar. Biol. 1994, 118, 53–65. [Google Scholar] [CrossRef]
- Yednock, B.K.; Neigel, J.E. An Investigation of Genetic Population Structure in Blue Crabs, Callinectes Sapidus, Using Nuclear Gene Sequences. Mar. Biol. 2014, 161, 871–886. [Google Scholar] [CrossRef]
- Macedo, D.; Caballero, I.; Mateos, M.; Leblois, R.; McCay, S.; Hurtado, L.A. Population Genetics and Historical Demographic Inferences of the Blue Crab Callinectes Sapidus in the US Based on Microsatellites. PeerJ 2019, 2019, e7780. [Google Scholar] [CrossRef] [Green Version]
- Plough, L. Population Genomic Analysis of the Blue Crab Callinectes Sapidus Using Genotyping-By-Sequencing. J. Shellfish Res. 2017, 36, 249–261. [Google Scholar] [CrossRef]
- Windsor, A.M.; Moore, M.K.; Warner, K.A.; Stadig, S.R.; Deeds, J.R. Evaluation of Variation within the Barcode Region of Cytochrome c Oxidase I (COI) for the Detection of Commercial Callinectes Sapidus Rathbun, 1896 (Blue Crab) Products of Non-US Origin. PeerJ 2019, 7, e7827. [Google Scholar] [CrossRef] [Green Version]
- Johnson, P.T.; Bodammer, J.E. A Disease of the Blue Crab, Callinectes Sapidus, of Possible Viral Etiology. J. Invertebr. Pathol. 1975, 26, 141–143. [Google Scholar] [CrossRef]
- Johnson, P.T. A Viral Disease of the Blue Crab, Callinectes Sapidus: Histopathology and Differential Diagnosis. J. Invertebr. Pathol. 1977, 29, 201–209. [Google Scholar] [CrossRef]
- Moya, A.; Elena, S.F.; Bracho, A.; Miralles, R.; Barrio, E. The Evolution of RNA Viruses: A Population Genetics View. Proc. Natl. Acad. Sci. USA 2000, 97, 6967–6973. [Google Scholar] [CrossRef] [Green Version]
- Walton, A.; Montanie, H.; Arcier, J.M.; Smith, V.J.; Bonami, J.R. Construction of a Gene Probe for Detection of P Virus (Reoviridae) in a Marine Decapod. J. Virol. Methods 1999, 81, 183–192. [Google Scholar] [CrossRef]
- Zhao, M.; dos Santos Tavares, C.P.; Schott, E.J. Diversity and Classification of Reoviruses in Crustaceans: A Proposal. J. Invertebr. Pathol. 2021, 182, 107568. [Google Scholar] [CrossRef] [PubMed]
- Flowers, E.M.; Bachvaroff, T.R.; Warg, J.V.; Neill, J.D.; Killian, M.L.; Vinagre, A.S.; Brown, S.; Almeida, A.S.E.; Schott, E.J. Genome Sequence Analysis of CsRV1: A Pathogenic Reovirus That Infects the Blue Crab Callinectes Sapidus Across Its Trans-Hemispheric Range. Front. Microbiol. 2016, 7, 126. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Ezpeleta, N.; Teijeiro, S.; Forget, L.; Burger, G.; Lang, B.F. Construction of CDNA Libraries: Focus on Protists and Fungi. Methods Mol. Biol. 2009, 533, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Flowers, E.M.; Schott, E.J. Near-Complete Sequence of a Highly Divergent Reovirus Genome Recovered from Callinectes Sapidus. Microbiol. Resour. Announc. 2021, 10, e01278-20. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR Method for MinION and Illumina Sequencing of Zika and Other Virus Genomes Directly from Clinical Samples. Nat. Protoc. 2017, 12, 1261–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation Software for the Fast Assembly of Multi-Gene Datasets with Character Set and Codon Information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Ma, Y.; Hu, Y. Near-Full-Length Genome Sequence of a Novel Reovirus from the Chinese Mitten Crab, Eriocheir sinensis. Genome Announc. 2015, 3, e00447-15. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, T.G. IcyTree: Rapid Browser-Based Visualization for Phylogenetic Trees and Networks. Bioinformatics 2017, 33, 2392–2394. [Google Scholar] [CrossRef] [Green Version]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. Package Version 1.0.7. R Package Version 2020, 1, 337–354. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.X.; Li, W.H. Statistical Tests of Neutrality of Mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef]
- Fu, Y.-X. Statistical Tests of Neutrality of Mutations Against Population Growth, Hitchhiking and Background Selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Akey, J.M. Genomic Insights into Positive Selection. Trends Genet. 2006, 22, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Harpending, H.C. Signature of Ancient Population Growth in a Low-Resolution Mitochondrial DNA Mismatch Distribution. Hum. Biol. 1994, 66, 591–600. [Google Scholar]
- Excoffier, L.; Lischer, H.E.L. Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.R.; Harpending, H. Population Growth Makes Waves in the Distribution of Pairwise Genetic Differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar] [CrossRef]
- Dray, S.; Dufour, A.-B. The Ade4 Package: Implementing the Duality Diagram for Ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Grenfell, B.T.; Pybus, O.G.; Gog, J.R.; Wood, J.L.N.; Daly, J.M.; Mumford, J.A.; Holmes, E.C. Unifying the Epidemiological and Evolutionary Dynamics of Pathogens. Science 2004, 303, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Neigel, J.; Plouviez, S.; Sullivan, T.; Yednock, B.; Rashbrook, V. Genetic Population Structure in Blue Crabs (Callinectes Sapidus): High Resolution Population Genomics of a High Gene Flow Species. Authorea (preprint). 2020. [Google Scholar] [CrossRef]
- Giachini Tosetto, E.; Bertrand, A.; Neumann-Leitão, S.; Nogueira Júnior, M. The Amazon River Plume, a Barrier to Animal Dispersal in the Western Tropical Atlantic. Sci. Rep. 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Sarver, S.K.; Silberman, J.D.; Walsh, P.J. Mitochondrial DNA Sequence Evidence Supporting the Recognition of Two Subspecies or Species of the Florida Spiny Lobster Panulirus argus. J. Crustacean Biol. 1998, 18, 177–186. [Google Scholar] [CrossRef]
- GIRALDES, B.W.; SMYTH, D.M. Recognizing Panulirus Meripurpuratus Sp. Nov. (Decapoda: Palinuridae) in Brazil—Systematic and Biogeographic Overview of Panulirus Species in the Atlantic Ocean. Zootaxa 2016, 4107, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goes, J.I.; do Rosario Gomes, H.; Chekalyuk, A.M.; Carpenter, E.J.; Montoya, J.P.; Coles, V.J.; Yager, P.L.; Berelson, W.M.; Capone, D.G.; Foster, R.A.; et al. Influence of the Amazon River Discharge on the Biogeography of Phytoplankton Communities in the Western Tropical North Atlantic. Prog. Oceanogr. 2014, 120, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Posey, M.H.; Alphin, T.D.; Harwell, H.; Allen, B. Importance of Low Salinity Areas for Juvenile Blue Crabs, Callinectes Sapidus Rathbun, in River-Dominated Estuaries of Southeastern United States. J. Exp. Mar. Biol. Ecol. 2005, 319, 81–100. [Google Scholar] [CrossRef]
- Cowen, R.K.; Sponaugle, S. Larval Dispersal and Marine Population Connectivity. Ann. Rev. Mar. Sci. 2009, 1, 443–466. [Google Scholar] [CrossRef] [Green Version]
- White, C.; Selkoe, K.A.; Watson, J.; Siegel, D.A.; Zacherl, D.C.; Toonen, R.J. Ocean Currents Help Explain Population Genetic Structure. Proc. R. Soc. B Biol. Sci. 2010, 277, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Criales, M.M.; Chérubin, L.; Gandy, R.; Garavelli, L.; Ghannami, M.A.; Crowley, C. Blue Crab Larval Dispersal Highlights Population Connectivity and Implications for Fishery Management. Mar. Ecol. Prog. Ser. 2019, 625, 53–70. [Google Scholar] [CrossRef] [Green Version]
- Britto, F.B.; Schmidt, A.J.; Carvalho, A.M.F.; Vasconcelos, C.C.M.P.; Farias, A.M.; Bentzen, P.; Diniz, F.M. Population Connectivity and Larval Dispersal of the Exploited Mangrove Crab Ucides Cordatus along the Brazilian Coast. PeerJ 2018, 6, e4702. [Google Scholar] [CrossRef] [Green Version]
- Lacerda, A.L.F.; Kersanach, R.; Cortinhas, M.C.S.; Prata, P.F.S.; Dumont, L.F.C.; Proietti, M.C.; Maggioni, R.; D’Incao, F. High Connectivity among Blue Crab (Callinectes Sapidus) Populations in the Western South Atlantic. PLoS ONE 2016, 11, e0153124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthelemy-Okazaki, N.J.; Okazaki, R.K. Population Genetics of the Blue Crab Callinectes Sapidus from the Northwestern Gulf of Mexico. Gulf Mex. Sci. 1997, 15, 4. [Google Scholar] [CrossRef]
- Cowen, R.K.; Paris, C.B.; Srinivasan, A. Scaling of Connectivity in Marine Populations. Science 2006, 311, 522–527. [Google Scholar] [CrossRef] [Green Version]
- Truelove, N.K.; Kough, A.S.; Behringer, D.C.; Paris, C.B.; Box, S.J.; Preziosi, R.F.; Butler, M.J. Biophysical Connectivity Explains Population Genetic Structure in a Highly Dispersive Marine Species. Coral Reefs 2017, 36, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Krkošek, M.; Gottesfeld, A.; Proctor, B.; Rolston, D.; Carr-Harris, C.; Lewis, M.A. Effects of Host Migration, Diversity and Aquaculture on Sea Lice Threats to Pacific Salmon Populations. Proc. R. Soc. B Biol. Sci. 2007, 274, 3141–3149. [Google Scholar] [CrossRef] [Green Version]
- McMillen-Jackson, A.L.; Bert, T.M. Mitochondrial DNA Variation and Population Genetic Structure of the Blue Crab Callinectes Sapidus in the Eastern United States. Mar. Biol. 2004, 145, 769–777. [Google Scholar] [CrossRef]
- Whittington, R.J.; Paul-Pont, I.; Evans, O.; Hick, P.; Dhand, N.K. Counting the Dead to Determine the Source and Transmission of the Marine Herpesvirus OsHV-1 in Crassostrea gigas. Vet. Res. 2018, 49, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hines, A.H.; Johnson, E.G.; Darnell, M.Z.; Rittschof, D.; Miller, T.J.; Bauer, L.J.; Rodgers, P.; Aguilar, R. Predicting Effects of Climate Change on Blue Crabs in Chesapeake Bay. In Proceedings of the Biology and Management of Exploited Crab Populations under Climate Change, Alaska Sea Grant, University of Alaska Fairbanks, April 27 2011; pp. 109–127. [Google Scholar]
- Japaud, A.; Bouchon, C.; Magalon, H.; Fauvelot, C. Geographic Distances and Ocean Currents Influence Caribbean Acropora Palmata Population Connectivity in the Lesser Antilles. Conserv. Genet. 2019, 20, 447–466. [Google Scholar] [CrossRef]
- Griffiths, S.M.; Butler, M.J.; Behringer, D.C.; Pérez, T.; Preziosi, R.F. Oceanographic Features and Limited Dispersal Shape the Population Genetic Structure of the Vase Sponge Ircinia Campana in the Greater Caribbean. Heredity 2021, 126, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Maan, S.; Maan, N.S.; Nomikou, K.; Veronesi, E.; Bachanek-Bankowska, K.; Belaganahalli, M.N.; Attoui, H.; Mertens, P.P.C. Complete Genome Characterisation of a Novel 26th Bluetongue Virus Serotype from Kuwait. PLoS ONE 2011, 6, e26147. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.S. The Savory Swimmer Swims North: A Northern Range Extension of the Blue Crab Callinectes Sapidus? J. Crustac. Biol. 2015, 35, 105–110. [Google Scholar] [CrossRef] [Green Version]
- Worobey, M.; Holmes, E.C. Evolutionary Aspects of Recombination in RNA Viruses. J. Gen. Virol. 1999, 80, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.M.; Nelson, M.I.; Turner, P.E.; Patton, J.T. Reassortment in Segmented RNA Viruses: Mechanisms and Outcomes. Nat. Rev. Microbiol. 2016, 14, 448–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svinti, V.; Cotton, J.A.; McInerney, J.O. New Approaches for Unravelling Reassortment Pathways. BMC Evol. Biol. 2013, 13, 1. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Latitude | Longitude | Collection Year | Seg9 Genome ID | Concatenated Genome ID |
|---|---|---|---|---|---|
| US Atlantic coast | |||||
| Westport River, MA | 41.5118° N | 71.0929° W | 2018 | MA 2018 #1–3 | MA 2018 #1;2 |
| Long Island, NY | 40.9395° N | 72.2304° W | 2011–2021 | NY 2018 #1–6 NY 2020 #1–4 | NY 2018 #4 NY 2020 #1 |
| Delaware Bay, DE | 38.9108° N | 75.5277° W | 2017 | DE 2017 #1–6 | DE 2017 #2;4 |
| Rhode River, MD | 38.8648° N | 76.5146° W | 2006–2020 | MD 2020 #1–2 | MD 2020 #1 |
| Chesapeake Bay, VA Albemarle Sound, NC Jacksonville, FL Gulf of Mexico Port Aransas, TX Dulac, LA Florida Keys, FLK Caribbean Sea Dominican Republic, DR Puerto Rico, PR Trinidad and Tobago, T&T South America Rio Grande do Sul, BR Uruguay, UY | 38.0214° N 33.8772° N 30.3322° N 27.8006° N 29.3888° N 24.8234° N 18.4511° N 18.4508° N 10.4633° N 30.0346° S 34.6285° S | 76.3524° W 76.1248° W 81.6557° W 97.3964° W 90.7140° W 80.8122° W 69.2133° W 65.9801° W 61.4836° W 51.2177° W 54.2921° W | 2018 2019 2018 2017 2019 2018 2018 2015 2017 2015 2019 | VA 2018 #1–2 NC 2019 #1–3 FL 2018 #1–5 TX 2017 #1–4 LA 2019 #1–11 FLK 2018 #1 DR 2018 #1 PR 2015 #1–2 T&T 2017 #1 BR 2015 #1–9 UY 2019 #1–6 | VA 2018 #1;2 NC 2019 #3 FL 2018 #2 TX 2017 #2 LA 2019 #1 FLK 2018 #1 NA NA T&T 2017 #1 BR 2015 #3;4;7;8 UY 2019 #1;5;6 |
| Dataset (Individual Number) | Nt ID% (Min–Max) | AA ID% (Min–Max) | H | Hd | π | ω (dN/dS) | θ |
|---|---|---|---|---|---|---|---|
| USAC + GoM + CAR + SAmer (96) | 95.73–100 | 94.31–100 | 67 | 0.98596 | 0.0140 | 0.17 | 0.029 |
| USAC (61) | 98.58–100 | 97.86–100 | 44 | 0.98251 | 0.0078 | 0.10 | 0.014 |
| GoM (16) | 96.44–100 | 94.66–100 | 12 | 0.94167 | 0.0135 | 0.29 | 0.019 |
| CAR (4) | 96.09–100 | 96.44–100 | 4 | 1.00000 | 0.0172 | 0.15 | 0.016 |
| SAmer (15) | 99.29–100 | 98.93–100 | 8 | 0.83810 | 0.0037 | 0.14 | 0.004 |
| Segment | Length (nt) | N of Sequences | % Nt ID (Min–Max) | % AA ID (Min–Max) | π | ω (dN/dS) | s | k | θ |
|---|---|---|---|---|---|---|---|---|---|
| Seg1 Seg2 Seg3 Seg4 Seg5 Seg6 Seg7 Seg8 Seg9 Seg10 Seg11 Seg12 | 4239 2337 2220 1959 1824 1620 1260 897 972 1032 612 837 | 23 23 23 23 23 23 23 23 23 23 23 23 | 97.3–99.7 97.1–99.9 97.6–99.9 96.8–99.9 96.2–100 97.3–99.9 96.3–100 96.4–100 96.1–100 95.2–100 97.7–100 97.6–100 | 97.5–99.9 97.1–99.0 97.4–100 95.7–99.9 95.2–100 95.9–100 94.3–100 93.6–100 93.8–100 90.7–100 97.0–100 96.4–100 | 0.014 0.012 0.012 0.015 0.016 0.017 0.021 0.018 0.018 0.021 0.010 0.011 | 0.11 0.12 0.08 0.19 0.15 0.21 0.30 0.40 0.18 0.37 0.08 0.14 | 352 207 163 172 170 140 126 74 87 125 42 63 | 60.23 28.95 26.99 30.15 28.89 28.15 26.81 16.24 17.47 22.29 5.68 9.04 | 0.023 0.025 0.020 0.025 0.027 0.025 0.031 0.025 0.026 0.035 0.019 0.021 |
| Segment | N of Sequences | SSD a | HRI a | Tajima’s D | Fu and Li’s D | Fu and Li’s F | Fu’s Fs d |
|---|---|---|---|---|---|---|---|
| Seg1 Seg2 Seg3 Seg4 Seg5 Seg6 Seg7 Seg8 Seg9 Seg10 Seg11 Seg12 | 23 23 23 23 23 23 23 23 23 23 23 23 | 0.0056 0.0051 0.0042 0.0060 0.0181 0.0039 0.0055 0.0133 0.0078 0.0079 0.0050 0.0049 | 0.0061 0.0091 0.0070 0.0122 0.0080 0.0098 0.0094 0.0102 0.0061 0.0095 0.0194 0.0071 | −1.58 −2.03 b −1.61 −1.54 −1.60 −1.18 −1.18 −1.07 −1.22 −1.57 −1.94 −1.89 | −2.60 b −3.14 c −2.63 b −2.54 b −2.28 −1.87 −1.78 −1.30 −2.04 −1.95 −3.07 c −2.54 b | −2.68 b −3.28 c −2.71 b −2.61 b −2.43 −1.94 −1.87 −1.44 −2.09 −2.15 −3.19 c −2.74 b | −3.73 −7.08 −7.50 −6.84 −4.81 −7.24 −5.19 −8.36 −5.83 −6.15 −5.70 −6.95 |
| Segment | Subpopulation 1 | Subpopulation 2 | FST * |
|---|---|---|---|
| Seg9 | USAC (61) | SAmer (15) | 0.69468 |
| (All regions) | USAC (61) | GoM (16) | 0.16164 |
| USAC (61) | CAR (4) | 0.66668 | |
| GoM (16) | CAR (4) | 0.51389 | |
| GoM (16) | SAmer (15) | 0.63118 | |
| CAR (4) | SAmer (15) | 0.62649 | |
| Seg9 (Within US) | N. Atl (25) N. Atl (25) N. Atl(25) M. Atl (31) M. Atl (31) S. Atl (5) | M. Atl (31) S. Atl (5) GoM (16) S. Atl (5) GoM (16) GoM (16) | 0.22894 0.57072 0.25073 0.47729 0.14819 0.35522 |
| Segment | Source of Variance | d.f. | Sum of Squares | Variance Component | Percentage of Variation | AMOVA Statistics | p-Value |
|---|---|---|---|---|---|---|---|
| Seg1 (All regions) | Among populations Within populations Total | 2 20 22 | 266.880 395.686 662.565 | 20.58327 Va 19.78429 Vb 40.36756 | 50.99 49.01 | 0.50990 | 0.00 |
| Seg8 | Among populations | 2 | 84.519 | 6.80089 Va | 59.10 | 0.59099 | 0.00 |
| (All regions) | Within populations | 20 | 94.133 | 4.70667 Vb | 40.90 | ||
| Total | 20 | 178.652 | 11.50756 | ||||
| Seg9 | Among populations | 2 | 212.912 | 6.47360 Va | 63.29 | 0.63290 | 0.00 |
| (All regions) | Within populations | 93 | 349.203 | 3.75487 Vb | 36.71 | ||
| Total | 95 | 562.115 | 10.22847 | ||||
| Seg9 | Among populations | 3 | 71.109 | 1.17949 Va | 26.76 | 0.26764 | 0.00 |
| (Within the US) | Within populations | 72 | 232.378 | 3.22747 Vb | 73.24 | ||
| Total | 75 | 303.487 | 4.40696 | ||||
| Seg9 | Among populations | 1 | 3.544 | 0.29582 Va | 17.30 | 0.17296 | 0.05 |
| (Within the SAmer) | Within populations | 13 | 18.389 | 1.41453 Vb | 82.70 | ||
| Total | 14 | 21.933 | 1.71035 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Plough, L.V.; Behringer, D.C.; Bojko, J.; Kough, A.S.; Alper, N.W.; Xu, L.; Schott, E.J. Cross-Hemispheric Genetic Diversity and Spatial Genetic Structure of Callinectes sapidus Reovirus 1 (CsRV1). Viruses 2023, 15, 563. https://doi.org/10.3390/v15020563
Zhao M, Plough LV, Behringer DC, Bojko J, Kough AS, Alper NW, Xu L, Schott EJ. Cross-Hemispheric Genetic Diversity and Spatial Genetic Structure of Callinectes sapidus Reovirus 1 (CsRV1). Viruses. 2023; 15(2):563. https://doi.org/10.3390/v15020563
Chicago/Turabian StyleZhao, Mingli, Louis V. Plough, Donald C. Behringer, Jamie Bojko, Andrew S. Kough, Nathaniel W. Alper, Lan Xu, and Eric J. Schott. 2023. "Cross-Hemispheric Genetic Diversity and Spatial Genetic Structure of Callinectes sapidus Reovirus 1 (CsRV1)" Viruses 15, no. 2: 563. https://doi.org/10.3390/v15020563