
Therapeutic Targeting of Inflammation and Virus Simultaneously Ameliorates Influenza Pneumonia and Protects from Morbidity and Mortality
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Ethics Statement
2.2. Mice
2.3. Cell Lines and Viruses
2.4. Virus Infection, Animal Weights, and Clinical Scores
2.5. Drug Treatments
2.6. Plaque Assay for Virus Quantification
2.7. TCID50 Assay for Virus Quantification
2.8. Lung Histopathological Examination
2.9. RNA Extraction, cDNA Generation, and Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.10. Statistical Analysis
3. Results
3.1. Etanercept Improves Clinical Signs and Reduces Lung Pathology without Affecting IAV Load in WT and TM Mice but TNF Deficiency Exacerbates Lung Pathology
3.2. Etanercept Combined with a Standard Dose of Oseltamivir (40 mg/kg) Daily Treatment Reduces Morbidity and Lung Pathology but Has No Effect on Viral Load
3.3. Combined Treatment with Etanercept and High Dose Oseltamivir Reduces Morbidity, Lung Viral Load, and Pathology in IAV-Infected Mice
3.4. Combined Treatment with Etanercept and High Dose Oseltamivir Reduces IAV Infection-Induced Lung Pathology Effectively in WT and TM Mice
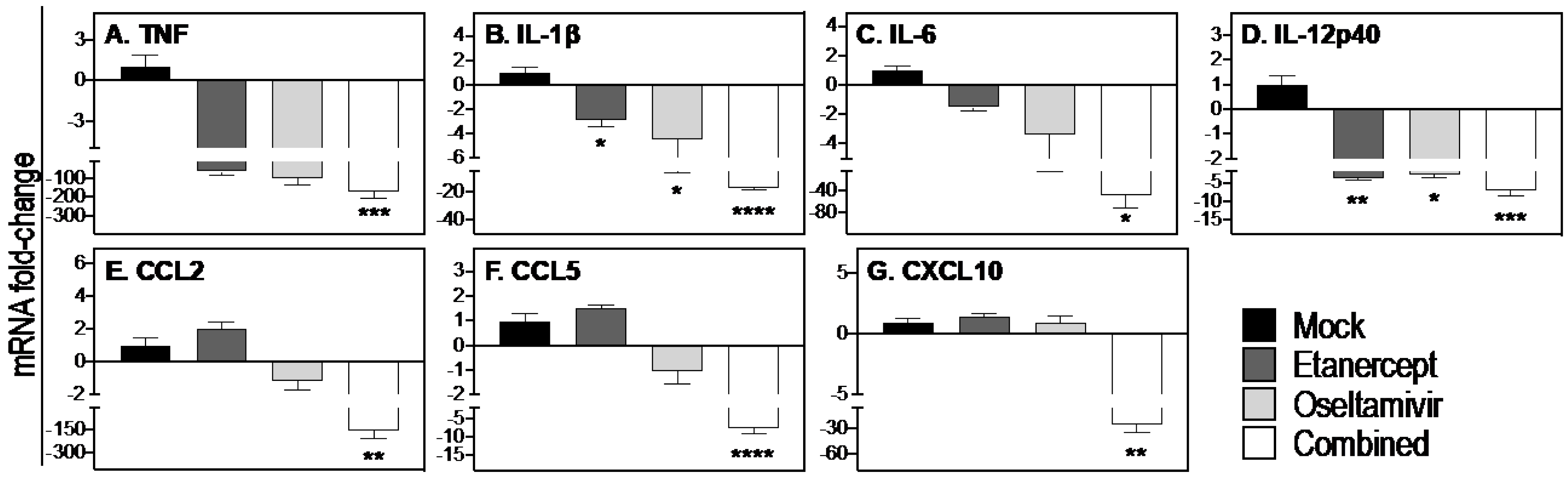
3.5. Combined Daily Treatment with Etanercept and High Dose Oseltamivir Reduces Expression of Inflammatory Cytokines and Chemokines
3.6. Combined Daily Treatment with Etanercept and High Dose Oseltamivir Protects Mice from Lethal IAV Infection
3.7. STAT3 Inhibitor in Combination with Oseltamivir Reduces Lung Viral Load and Improves Lung Pathology and Morbidity Associated with Severe IAV Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.; Chau, T.N.; Hoang, D.M.; Chau, N.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Rello, J.; Pop-Vicas, A. Clinical review: Primary influenza viral pneumonia. Crit. Care 2009, 13, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rello, J.; Rodríguez, A.; Ibañez, P.; Socias, L.; Cebrian, J.; Marques, A.; Guerrero, J.; Ruiz-Santana, S.; Marquez, E.; Del Nogal-Saez, F.; et al. Intensive care adult patients with severe respiratory failure caused by Influenza A (H1N1)v in Spain. Crit. Care 2009, 13, R148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothberg, M.B.; Haessler, S.D.; Brown, R.B. Complications of viral influenza. Am. J. Med. 2008, 121, 258–264. [Google Scholar] [CrossRef]
- Jefferson, T.; Jones, M.A.; Doshi, P.; Del Mar, C.B.; Hama, R.; Thompson, M.J.; Spencer, E.A.; Onakpoya, I.; Mahtani, K.R.; Nunan, D.; et al. Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children. Cochrane Database Syst. Rev. 2014, 2014, Cd008965. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Chung, J.W.; Kim, T.; Park, K.H.; Lee, M.S.; Kwak, Y.G. Late diagnosis of influenza in adult patients during a seasonal outbreak. Korean J. Intern. Med. 2018, 33, 391–396. [Google Scholar] [CrossRef] [Green Version]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [Green Version]
- La Gruta, N.L.; Kedzierska, K.; Stambas, J.; Doherty, P.C. A question of self-preservation: Immunopathology in influenza virus infection. Immunol. Cell Biol. 2007, 85, 85–92. [Google Scholar] [CrossRef]
- Kobasa, D.; Jones, S.M.; Shinya, K.; Kash, J.C.; Copps, J.; Ebihara, H.; Hatta, Y.; Kim, J.H.; Halfmann, P.; Hatta, M.; et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 2007, 445, 319–323. [Google Scholar] [CrossRef]
- Oshansky, C.M.; Gartland, A.J.; Wong, S.-S.; Jeevan, T.; Wang, D.; Roddam, P.L.; Caniza, M.A.; Hertz, T.; DeVincenzo, J.P.; Webby, R.J.; et al. Mucosal Immune Responses Predict Clinical Outcomes during Influenza Infection Independently of Age and Viral Load. Am. J. Respir. Crit. Care Med. 2014, 189, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Schall, T.J.; Bacon, K.B. Chemokines, leukocyte trafficking, and inflammation. Curr. Opin. Immunol. 1994, 6, 865–873. [Google Scholar] [CrossRef]
- Alon, R.; Sportiello, M.; Kozlovski, S.; Kumar, A.; Reilly, E.C.; Zarbock, A.; Garbi, N.; Topham, D.J. Leukocyte trafficking to the lungs and beyond: Lessons from influenza for COVID-19. Nat. Rev. Immunol. 2021, 21, 49–64. [Google Scholar] [CrossRef]
- Kudo, K.; Takasaki, J.; Manabe, T.; Uryu, H.; Yamada, R.; Kuroda, E.; Kobayashi, N.; Matsushita, T. Systemic corticosteroids and early administration of antiviral agents for pneumonia with acute wheezing due to influenza A(H1N1)pdm09 in Japan. PLoS ONE 2012, 7, e32280. [Google Scholar] [CrossRef]
- Zheng, B.-J.; Chan, K.-W.; Lin, Y.-P.; Zhao, G.-Y.; Chan, C.; Zhang, H.-J.; Chen, H.-L.; Wong, S.S.Y.; Lau, S.K.P.; Woo, P.C.Y.; et al. Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc. Natl. Acad. Sci. USA 2008, 105, 8091–8096. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Zhou, W.; Huang, H.; Zhu, H.; Zhou, P.; Zhu, H.; Ju, D. Inhibition of the inflammatory cytokine tumor necrosis factor-alpha with etanercept provides protection against lethal H1N1 influenza infection in mice. Crit. Care 2013, 17, R301. [Google Scholar] [CrossRef] [Green Version]
- Pandey, P.; Karupiah, G. Targeting tumour necrosis factor to ameliorate viral pneumonia. FEBS J. 2021, 289, 883–900. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, Y.H.; Yang, Z.Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016, 13, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Peper, R.L.; Van Campen, H. Tumor necrosis factor as a mediator of inflammation in influenza A viral pneumonia. Microb. Pathog. 1995, 19, 175–183. [Google Scholar] [CrossRef]
- Belisle, S.E.; Tisoncik, J.R.; Korth, M.J.; Carter, V.S.; Proll, S.C.; Swayne, D.E.; Pantin-Jackwood, M.; Tumpey, T.M.; Katze, M.G. Genomic profiling of tumor necrosis factor alpha (TNF-alpha) receptor and interleukin-1 receptor knockout mice reveals a link between TNF-alpha signaling and increased severity of 1918 pandemic influenza virus infection. J. Virol. 2010, 84, 12576–12588. [Google Scholar] [CrossRef] [Green Version]
- Hayden, F.G.; Fritz, R.; Lobo, M.C.; Alvord, W.; Strober, W.; Straus, S.E. Local and systemic cytokine responses during experimental human influenza A virus infection. Relation to symptom formation and host defense. J. Clin. Invest. 1998, 101, 643–649. [Google Scholar] [CrossRef]
- Pires, B.R.B.; Silva, R.; Ferreira, G.M.; Abdelhay, E. NF-kappaB: Two Sides of the Same Coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, A.D.; Hunt, N.H.; Wanigasekara, Y.; Bloomfield, G.; Wallach, D.; Roufogalis, B.D.; Chaudhri, G. A casein kinase I motif present in the cytoplasmic domain of members of the tumour necrosis factor ligand family is implicated in ‘reverse signalling’. EMBO J. 1999, 18, 2119–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissner, G.; Kolch, W.; Scheurich, P. Ligands working as receptors: Reverse signaling by members of the TNF superfamily enhance the plasticity of the immune system. Cytokine Growth Factor Rev. 2004, 15, 353–366. [Google Scholar] [CrossRef]
- DTB. Oseltamivir for influenza. Drug. Ther. Bull. 2002, 40, 89–91. [Google Scholar] [CrossRef]
- CDC. Influenza Antiviral Medications: Summary for Clinicians. Available online: https://www.cdc.gov/flu/professionals/antivirals/summary-clinicians.htm (accessed on 20 September 2022).
- De Clercq, E. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discov. 2006, 5, 1015–1025. [Google Scholar] [CrossRef]
- Ramiro, S.; Radner, H.; van der Heijde, D.; van Tubergen, A.; Buchbinder, R.; Aletaha, D.; Landewé, R.B. Combination therapy for pain management in inflammatory arthritis (rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, other spondyloarthritis). Cochrane Database Syst. Rev. 2011, Cd008886. [Google Scholar] [CrossRef]
- Tuazon Kels, M.J.; Ng, E.; Al Rumaih, Z.; Pandey, P.; Ruuls, S.R.; Korner, H.; Newsome, T.P.; Chaudhri, G.; Karupiah, G. TNF deficiency dysregulates inflammatory cytokine production, leading to lung pathology and death during respiratory poxvirus infection. Proc. Natl. Acad. Sci. USA 2020, 117, 15935–15946. [Google Scholar] [CrossRef]
- Al Rumaih, Z.; Kels, M.J.T.; Ng, E.; Pandey, P.; Pontejo, S.M.; Alejo, A.; Alcamí, A.; Chaudhri, G.; Karupiah, G. Poxvirus-encoded TNF receptor homolog dampens inflammation and protects from uncontrolled lung pathology during respiratory infection. Proc. Natl. Acad. Sci. USA 2020, 117, 26885–26894. [Google Scholar] [CrossRef]
- Körner, H.; Cook, M.; Riminton, D.S.; Lemckert, F.A.; Hoek, R.M.; Ledermann, B.; Köntgen, F.; de St Groth, B.F.; Sedgwick, J.D. Distinct roles for lymphotoxin-α and tumor necrosis factor in organogenesis and spatial organization of lymphoid tissue. Eur. J. Immunol. 1997, 27, 2600–2609. [Google Scholar] [CrossRef]
- Balish, A.L.; Katz, J.M.; Klimov, A.I. Influenza: Propagation, quantification, and storage. Curr. Protoc. Microbiol. 2013, 29, 15G. 11.11–15G. 11.24. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Kaymakcalan, Z.; Sakorafas, P.; Bose, S.; Scesney, S.; Xiong, L.; Hanzatian, D.K.; Salfeld, J.; Sasso, E.H. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin. Immunol. 2009, 131, 308–316. [Google Scholar] [CrossRef]
- Meusch, U.; Rossol, M.; Baerwald, C.; Hauschildt, S.; Wagner, U. Outside-to-inside signaling through transmembrane tumor necrosis factor reverses pathologic interleukin-1beta production and deficient apoptosis of rheumatoid arthritis monocytes. Arthritis Rheum. 2009, 60, 2612–2621. [Google Scholar] [CrossRef]
- Pandey, P.; Al Rumaih, Z.; Kels, M.J.T.; Ng, E.; Kc, R.; Chaudhri, G.; Karupiah, G. Targeting ectromelia virus and TNF/NF-κB or STAT3 signaling for effective treatment of viral pneumonia. Proc. Natl. Acad. Sci. USA 2022, 119, e2112725119. [Google Scholar] [CrossRef]
- Damjanovic, D.; Divangahi, M.; Kugathasan, K.; Small, C.L.; Zganiacz, A.; Brown, E.G.; Hogaboam, C.M.; Gauldie, J.; Xing, Z. Negative regulation of lung inflammation and immunopathology by TNF-α during acute influenza infection. Am. J. Pathol. 2011, 179, 2963–2976. [Google Scholar] [CrossRef]
- Dutkowski, R.; Thakrar, B.; Froehlich, E.; Suter, P.; Oo, C.; Ward, P. Safety and pharmacology of oseltamivir in clinical use. Drug Saf. 2003, 26, 787–801. [Google Scholar] [CrossRef]
- Ward, P.; Small, I.; Smith, J.; Suter, P.; Dutkowski, R. Oseltamivir (Tamiflu) and its potential for use in the event of an influenza pandemic. J. Antimicrob. Chemother. 2005, 55, i5–i21. [Google Scholar] [CrossRef] [Green Version]
- Hierholzer, J.C.; Killington, R.A. 2—Virus isolation and quantitation. In Virology Methods Manual; Mahy, B.W.J., Kangro, H.O., Eds.; Academic Press: London, UK, 1996; pp. 25–46. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Karupiah, G.; Panchanathan, V.; Sakala, I.G.; Chaudhri, G. Genetic resistance to smallpox: Lessons from mousepox. Novartis Found. Symp. 2007, 281, 129–136, discussion 136–140, 208–129. [Google Scholar] [CrossRef]
- Ross, P.J.; Seaton, A.; Foreman, H.M.; Morris Evans, W.H. Pulmonary calcification following smallpox handler’s lung. Thorax 1974, 29, 659–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Influenza Strategy 2019–2030; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Collaborators, G.I. Mortality, morbidity, and hospitalisations due to influenza lower respiratory tract infections, 2017: An analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2019, 7, 69–89. [Google Scholar] [CrossRef] [Green Version]
- Bray, M.; Martinez, M.; Smee, D.F.; Kefauver, D.; Thompson, E.; Huggins, J.W. Cidofovir protects mice against lethal aerosol or intranasal cowpox virus challenge. J. Infect. Dis. 2000, 181, 10–19. [Google Scholar] [CrossRef]
- Quenelle, D.C.; Collins, D.J.; Kern, E.R. Efficacy of multiple- or single-dose cidofovir against vaccinia and cowpox virus infections in mice. Antimicrob. Agents Chemother. 2003, 47, 3275–3280. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.T.; Grosenbach, D.W.; Brasel, T.L.; Baker, R.O.; Cawthon, A.G.; Reynolds, E.; Bailey, T.; Kuehl, P.J.; Sugita, V.; Agans, K.; et al. Effects of Treatment Delay on Efficacy of Tecovirimat Following Lethal Aerosol Monkeypox Virus Challenge in Cynomolgus Macaques. J. Infect. Dis. 2018, 218, 1490–1499. [Google Scholar] [CrossRef] [Green Version]
- Cernik, C.; Gallina, K.; Brodell, R.T. The Treatment of Herpes Simplex Infections: An Evidence-Based Review. Arch. Intern. Med. 2008, 168, 1137–1144. [Google Scholar] [CrossRef] [Green Version]
- Consortium, W.S.T. Repurposed Antiviral Drugs for Covid-19—Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2020, 384, 497–511. [Google Scholar] [CrossRef]
- Xu, T.; Qiao, J.; Zhao, L.; He, G.; Li, K.; Wang, J.; Tian, Y.; Wang, H. Effect of dexamethasone on acute respiratory distress syndrome induced by the H5N1 virus in mice. Eur. Respir. J. 2009, 33, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Salomon, R.; Hoffmann, E.; Webster, R.G. Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc. Natl. Acad. Sci. USA 2007, 104, 12479–12481. [Google Scholar] [CrossRef] [Green Version]
- Walsh, K.B.; Teijaro, J.R.; Wilker, P.R.; Jatzek, A.; Fremgen, D.M.; Das, S.C.; Watanabe, T.; Hatta, M.; Shinya, K.; Suresh, M.; et al. Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc. Natl. Acad. Sci. USA 2011, 108, 12018–12023. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.L.; Sweeney, S.; Kang, B.D.; Ramsburg, E.; Gunn, M.D. CCR2-antagonist prophylaxis reduces pulmonary immune pathology and markedly improves survival during influenza infection. J. Immunol. 2011, 186, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Moseley, C.E.; Webster, R.G.; Aldridge, J.R. Peroxisome proliferator-activated receptor and AMP-activated protein kinase agonists protect against lethal influenza virus challenge in mice. Influenza Other Respir. Viruses 2010, 4, 307–311. [Google Scholar] [CrossRef]
- Boon, A.C.; Finkelstein, D.; Zheng, M.; Liao, G.; Allard, J.; Klumpp, K.; Webster, R.; Peltz, G.; Webby, R.J. H5N1 influenza virus pathogenesis in genetically diverse mice is mediated at the level of viral load. mBio 2011, 2, e00171-11. [Google Scholar] [CrossRef] [Green Version]
- To, K.K.; Hung, I.F.; Li, I.W.; Lee, K.L.; Koo, C.K.; Yan, W.W.; Liu, R.; Ho, K.Y.; Chu, K.H.; Watt, C.L.; et al. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clin. Infect. Dis. 2010, 50, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Hussell, T.; Pennycook, A.; Openshaw, P.J. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur. J. Immunol. 2001, 31, 2566–2573. [Google Scholar] [CrossRef]
- Van Reeth, K.; Van Gucht, S.; Pensaert, M. Correlations between lung proinflammatory cytokine levels, virus replication, and disease after swine influenza virus challenge of vaccination-immune pigs. Viral Immunol. 2002, 15, 583–594. [Google Scholar] [CrossRef]
- Fritz, R.S.; Hayden, F.G.; Calfee, D.P.; Cass, L.M.; Peng, A.W.; Alvord, W.G.; Strober, W.; Straus, S.E. Nasal cytokine and chemokine responses in experimental influenza A virus infection: Results of a placebo-controlled trial of intravenous zanamivir treatment. J. Infect. Dis. 1999, 180, 586–593. [Google Scholar] [CrossRef]
- Silva, L.C.; Ortigosa, L.C.; Benard, G. Anti-TNF-α agents in the treatment of immune-mediated inflammatory diseases: Mechanisms of action and pitfalls. Immunotherapy 2010, 2, 817–833. [Google Scholar] [CrossRef]
- Smee, D.F.; Wong, M.H.; Bailey, K.W.; Sidwell, R.W. Activities of oseltamivir and ribavirin used alone and in combination against infections in mice with recent isolates of influenza A (H1N1) and B viruses. Antivir. Chem. Chemother. 2006, 17, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, J.G.; Albrecht, U.; Häussinger, D.; Heinrich, P.C.; Schaper, F. Hepatic acute phase proteins--regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-κB-dependent signaling. Eur. J. Cell Biol. 2012, 91, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-κB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Ji, Z.; He, L.; Regev, A.; Struhl, K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9453–9462. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Fajnzylber, J.; Regan, J.; Coxen, K.; Corry, H.; Wong, C.; Rosenthal, A.; Worrall, D.; Giguel, F.; Piechocka-Trocha, A.; Atyeo, C.; et al. SARS-CoV-2 viral load is associated with increased disease severity and mortality. Nat. Commun. 2020, 11, 5493. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Ryabkova, V.A.; Churilov, L.P.; Shoenfeld, Y. Influenza infection, SARS, MERS and COVID-19: Cytokine storm—The common denominator and the lessons to be learned. Clin. Immunol. 2021, 223, 108652. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, P.; Al Rumaih, Z.; Kels, M.J.T.; Ng, E.; Kc, R.; Malley, R.; Chaudhri, G.; Karupiah, G. Therapeutic Targeting of Inflammation and Virus Simultaneously Ameliorates Influenza Pneumonia and Protects from Morbidity and Mortality. Viruses 2023, 15, 318. https://doi.org/10.3390/v15020318
Pandey P, Al Rumaih Z, Kels MJT, Ng E, Kc R, Malley R, Chaudhri G, Karupiah G. Therapeutic Targeting of Inflammation and Virus Simultaneously Ameliorates Influenza Pneumonia and Protects from Morbidity and Mortality. Viruses. 2023; 15(2):318. https://doi.org/10.3390/v15020318
Chicago/Turabian StylePandey, Pratikshya, Zahrah Al Rumaih, Ma. Junaliah Tuazon Kels, Esther Ng, Rajendra Kc, Roslyn Malley, Geeta Chaudhri, and Gunasegaran Karupiah. 2023. "Therapeutic Targeting of Inflammation and Virus Simultaneously Ameliorates Influenza Pneumonia and Protects from Morbidity and Mortality" Viruses 15, no. 2: 318. https://doi.org/10.3390/v15020318