
Discovery of a Novel Intron in US10/US11/US12 of HSV-1 Strain 17


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Genome Sequence and RNA-Seq Data
2.2. Analysis of US10/US11/US12 Gene and Protein
2.3. Phylogenetic Analysis of HSV-1 Strains with Different US11 Gene Length
3. Results
3.1. Discovery of Novel Intron in US10/US11/US12 of HSV-1 Strain 17
3.2. Survey of the Intron Region of US11/US10/US12 in HSV-1 Strains
3.3. HSV-1 US11 and US10 Protein Structure
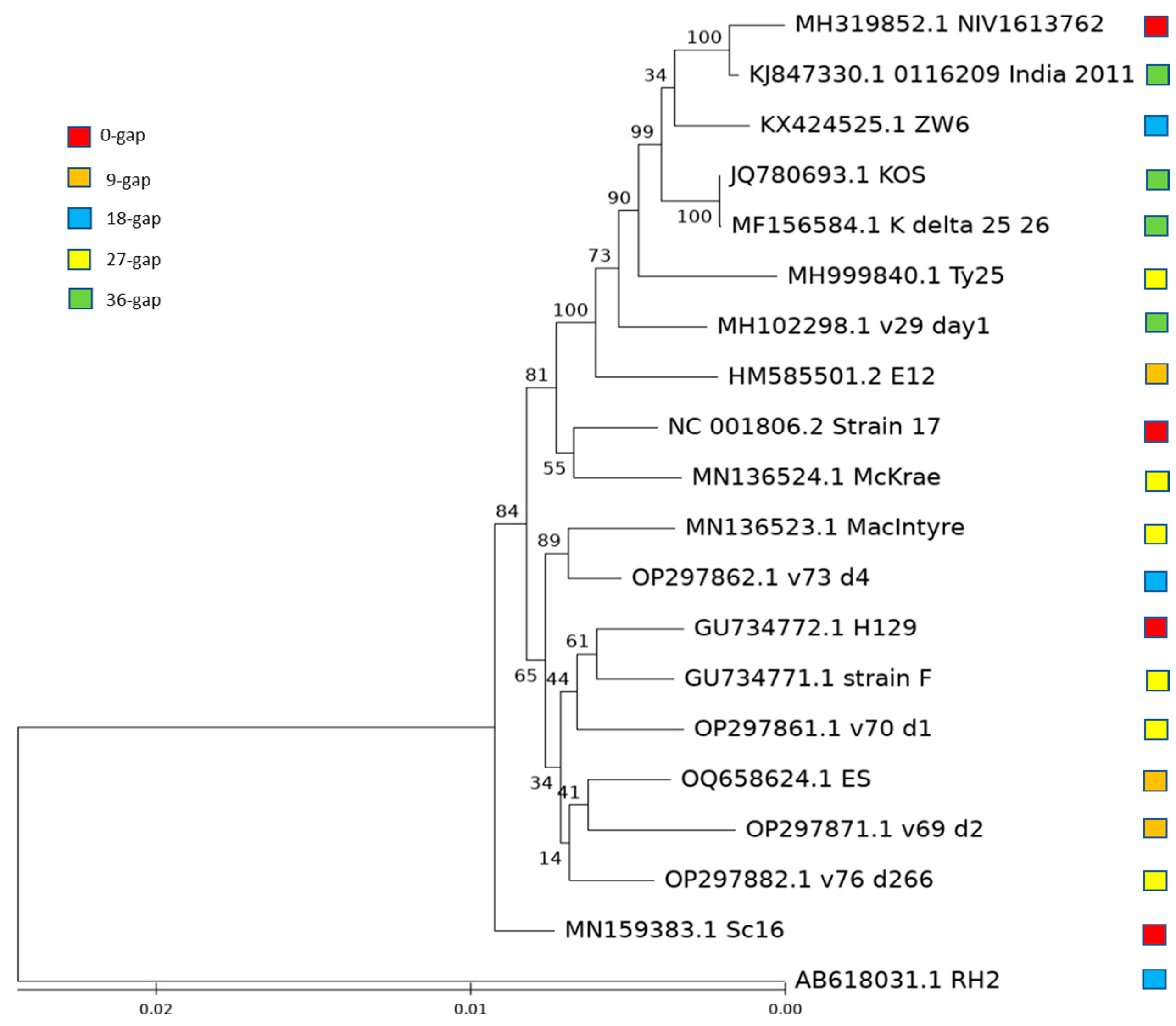
3.4. Evolution of HSV-1 with Different Length of US10/US11 Gene
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whitley, R.J.; Kimberlin, D.W.; Roizman, B. Herpes simplex viruses. Clin. Infect. Dis. 1998, 26, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Gatherer, D.; Depledge, D.P.; Hartley, C.A.; Szpara, M.L.; Vaz, P.K.; Benkő, M.; Brandt, C.R.; Bryant, N.A.; Dastjerdi, A.; Doszpoly, A.; et al. ICTV Virus Taxonomy Profile: Herpesviridae 2021. J. Gen. Virol. 2021, 102, 001673. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.I.; Bellamy, A.R.; Hook, E.W., 3rd; Levin, M.J.; Wald, A.; Ewell, M.G.; Wolff, P.A.; Deal, C.D.; Heineman, T.C.; Dubin, G.; et al. Epidemiology, clinical presentation, and antibody response to primary infection with herpes simplex virus type 1 and type 2 in young women. Clin. Infect. Dis. 2013, 56, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Gnann, J.W., Jr.; Whitley, R.J. Clinical Practice. Genital Herpes. N. Engl. J. Med. 2016, 375, 666–674. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J.; Dalrymple, M.A.; Davison, A.J.; Dolan, A.; Frame, M.C.; McNab, D.; Perry, L.J.; Scott, J.E.; Taylor, P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 1988, 69 Pt 7, 1531–1574. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J. Evolution of sexually transmitted and sexually transmissible human herpesviruses. Ann. N. Y. Acad. Sci. 2011, 1230, E37–E49. [Google Scholar] [CrossRef]
- Perry, L.J.; McGeoch, D.J. The DNA sequences of the long repeat region and adjoining parts of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 1988, 69 Pt 11, 2831–2846. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Dolan, A.; Donald, S.; Rixon, F.J. Sequence determination and genetic content of the short unique region in the genome of herpes simplex virus type 1. J. Mol. Biol. 1985, 181, 1–13. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Dolan, A.; Donald, S.; Brauer, D.H. Complete DNA sequence of the short repeat region in the genome of herpes simplex virus type 1. Nucleic Acids Res. 1986, 14, 1727–1745. [Google Scholar] [CrossRef]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef]
- Oehler, J.B.; Wright, H.; Stark, Z.; Mallett, A.J.; Schmitz, U. The application of long-read sequencing in clinical settings. Hum. Genom. 2023, 17, 73. [Google Scholar] [CrossRef] [PubMed]
- Enko, D.; Michaelis, S.; Schneider, C.; Schaflinger, E.; Baranyi, A.; Schnedl, W.J.; Muller, D.J. The Use of Next-Generation Sequencing in Pharmacogenomics. Clin. Lab. 2023, 69. [Google Scholar] [CrossRef] [PubMed]
- Carangelo, G.; Magi, A.; Semeraro, R. From multitude to singularity: An up-to-date overview of scRNA-seq data generation and analysis. Front. Genet. 2022, 13, 994069. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Hu, N.; Mo, L.; Zeng, Z.; Sun, J.; Hu, Y. Deep RNA Sequencing Reveals a Repertoire of Human Fibroblast Circular RNAs Associated with Cellular Responses to Herpes Simplex Virus 1 Infection. Cell. Physiol. Biochem. 2018, 47, 2031–2045. [Google Scholar] [CrossRef]
- Tombácz, D.; Csabai, Z.; Szűcs, A.; Balázs, Z.; Moldován, N.; Sharon, D.; Snyder, M.; Boldogkői, Z. Long-Read Isoform Sequencing Reveals a Hidden Complexity of the Transcriptional Landscape of Herpes Simplex Virus Type 1. Front. Microbiol. 2017, 8, 1079. [Google Scholar] [CrossRef]
- Tombácz, D.; Moldován, N.; Balázs, Z.; Gulyás, G.; Csabai, Z.; Boldogkői, M.; Snyder, M.; Boldogkői, Z. Multiple Long-Read Sequencing Survey of Herpes Simplex Virus Dynamic Transcriptome. Front. Genet. 2019, 10, 834. [Google Scholar] [CrossRef]
- Yamada, H.; Daikoku, T.; Yamashita, Y.; Jiang, Y.M.; Tsurumi, T.; Nishiyama, Y. The product of the US10 gene of herpes simplex virus type 1 is a capsid/tegument-associated phosphoprotein which copurifies with the nuclear matrix. J. Gen. Virol. 1997, 78 Pt 11, 2923–2931. [Google Scholar] [CrossRef]
- Johnson, P.A.; MacLean, C.; Marsden, H.S.; Dalziel, R.G.; Everett, R.D. The product of gene US11 of herpes simplex virus type 1 is expressed as a true late gene. J. Gen. Virol. 1986, 67 Pt 5, 871–883. [Google Scholar] [CrossRef]
- Roller, R.J.; Monk, L.L.; Stuart, D.; Roizman, B. Structure and function in the herpes simplex virus 1 RNA-binding protein U(s)11: Mapping of the domain required for ribosomal and nucleolar association and RNA binding in vitro. J. Virol. 1996, 70, 2842–2851. [Google Scholar] [CrossRef]
- Roller, R.J.; Roizman, B. The herpes simplex virus 1 RNA binding protein US11 is a virion component and associates with ribosomal 60S subunits. J. Virol. 1992, 66, 3624–3632. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.J.; Dodon, M.D.; Schaerer-Uthurralt, N.; Simonin, D.; Kindbeiter, K.; Gazzolo, L.; Madjar, J.J. Post-transcriptional transactivation of human retroviral envelope glycoprotein expression by herpes simplex virus Us11 protein. Nature 1996, 379, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Cassady, K.A.; Gross, M.; Roizman, B. The herpes simplex virus US11 protein effectively compensates for the gamma1 (34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J. Virol. 1998, 72, 8620–8626. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Arata, L.; Soler, E.; Gaume, X.; Couté, Y.; Hacot, S.; Callé, A.; Monier, K.; Epstein, A.L.; Sanchez, J.C.; et al. Nucleolin interacts with US11 protein of herpes simplex virus 1 and is involved in its trafficking. J. Virol. 2012, 86, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Benboudjema, L.; Mulvey, M.; Gao, Y.; Pimplikar, S.W.; Mohr, I. Association of the herpes simplex virus type 1 Us11 gene product with the cellular kinesin light-chain-related protein PAT1 results in the redistribution of both polypeptides. J. Virol. 2003, 77, 9192–9203. [Google Scholar] [CrossRef]
- Nouri, K.; Moll, J.M.; Milroy, L.G.; Hain, A.; Dvorsky, R.; Amin, E.; Lenders, M.; Nagel-Steger, L.; Howe, S.; Smits, S.H.; et al. Biophysical Characterization of Nucleophosmin Interactions with Human Immunodeficiency Virus Rev and Herpes Simplex Virus US11. PLoS ONE 2015, 10, e0143634. [Google Scholar] [CrossRef]
- Liu, X.; Matrenec, R.; Gack, M.U.; He, B. Disassembly of the TRIM23-TBK1 Complex by the Us11 Protein of Herpes Simplex Virus 1 Impairs Autophagy. J. Virol. 2019, 93, e00497-19. [Google Scholar] [CrossRef]
- Mangold, C.A.; Rathbun, M.M.; Renner, D.W.; Kuny, C.V.; Szpara, M.L. Viral infection of human neurons triggers strain-specific differences in host neuronal and viral transcriptomes. PLoS Pathog. 2021, 17, e1009441. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Gale, C.V.; Myers, R.; Tedder, R.S.; Williams, I.G.; Kellam, P. Development of a novel human immunodeficiency virus type 1 subtyping tool, Subtype Analyzer (STAR): Analysis of subtype distribution in London. AIDS Res. Hum. Retroviruses 2004, 20, 457–464. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Jiao, X.; Sui, H.; Goswami, S.; Sherman, B.T.; Fromont, C.; Caravaca, J.M.; Tran, B.; Imamichi, T. Complete Genome Sequence of Herpes Simplex Virus 2 Strain G. Viruses 2022, 14, 536. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Gilson, P.R.; Su, V.; Slamovits, C.H.; Reith, M.E.; Keeling, P.J.; McFadden, G.I. Complete nucleotide sequence of the chlorarachniophyte nucleomorph: Nature’s smallest nucleus. Proc. Natl. Acad. Sci. USA 2006, 103, 9566–9571. [Google Scholar] [CrossRef]
- Jackson, I.J. A reappraisal of non-consensus mRNA splice sites. Nucleic Acids Res. 1991, 19, 3795–3798. [Google Scholar] [CrossRef]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef]
- Umene, K.; Kawana, T. Divergence of reiterated sequences in a series of genital isolates of herpes simplex virus type 1 from individual patients. J. Gen. Virol. 2003, 84 Pt 4, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Umene, K.; Yoshida, M. Reiterated sequences of herpes simplex virus type 1 (HSV-1) genome can serve as physical markers for the differentiation of HSV-1 strains. Arch. Virol. 1989, 106, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Burset, M.; Seledtsov, I.A.; Solovyev, V.V. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000, 28, 4364–4375. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.M. Noncanonical GA and GG 5′ Intron Donor Splice Sites Are Common in the Copepod Eurytemora affinis. G3 2017, 7, 3967–3969. [Google Scholar] [CrossRef]
- Rixon, F.J.; McGeoch, D.J. A 3′ co-terminal family of mRNAs from the herpes simplex virus type 1 short region: Two overlapping reading frames encode unrelated polypeptide one of which has highly reiterated amino acid sequence. Nucleic Acids Res. 1984, 12, 2473–2487. [Google Scholar] [CrossRef]
- Schaerer-Uthurralt, N.; Erard, M.; Kindbeiter, K.; Madjar, J.J.; Diaz, J.J. Distinct domains in herpes simplex virus type 1 US11 protein mediate post-transcriptional transactivation of human T-lymphotropic virus type I envelope glycoprotein gene expression and specific binding to the Rex responsive element. J. Gen. Virol. 1998, 79 Pt 7, 1593–1602. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Kurachi, R.; Daikoku, T.; Umene, K. The US 9, 10, 11, and 12 genes of herpes simplex virus type 1 are of no importance for its neurovirulence and latency in mice. Virology 1993, 194, 419–423. [Google Scholar] [CrossRef]
- Mayr, C. What Are 3′ UTRs Doing? Cold Spring Harb. Perspect. Biol. 2019, 11, a034728. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Montgomerie, S.; Cruz, J.A.; Shrivastava, S.; Arndt, D.; Berjanskii, M.; Wishart, D.S. PROTEUS2: A web server for comprehensive protein structure prediction and structure-based annotation. Nucleic Acids Res. 2008, 36 (Suppl 2), W202–W209. [Google Scholar] [CrossRef]
- Høie, M.H.; Kiehl, E.N.; Petersen, B.; Nielsen, M.; Winther, O.; Nielsen, H.; Hallgren, J.; Marcatili, P. NetSurfP-3.0: Accurate and fast prediction of protein structural features by protein language models and deep learning. Nucleic Acids Res. 2022, 50, W510–W515. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32 Suppl. 2), W526–W531. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 2012, 80, 1715–1735. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, W.; Hao, M.; Qiu, J.; Sherman, B.T.; Imamichi, T. Discovery of a Novel Intron in US10/US11/US12 of HSV-1 Strain 17. Viruses 2023, 15, 2144. https://doi.org/10.3390/v15112144
Chang W, Hao M, Qiu J, Sherman BT, Imamichi T. Discovery of a Novel Intron in US10/US11/US12 of HSV-1 Strain 17. Viruses. 2023; 15(11):2144. https://doi.org/10.3390/v15112144
Chicago/Turabian StyleChang, Weizhong, Ming Hao, Ju Qiu, Brad T. Sherman, and Tomozumi Imamichi. 2023. "Discovery of a Novel Intron in US10/US11/US12 of HSV-1 Strain 17" Viruses 15, no. 11: 2144. https://doi.org/10.3390/v15112144