
Strategies in the Design and Development of Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs)

 ,
, 
Abstract
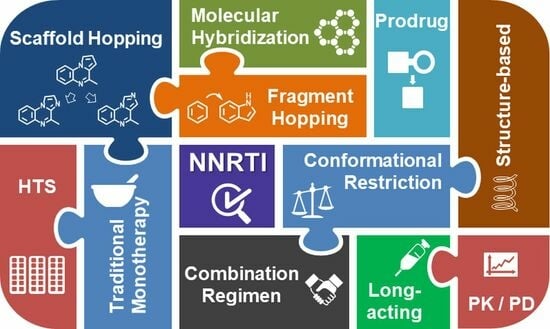
:
1. Introduction
2. Strategies for the Design and Development of NNRTIs
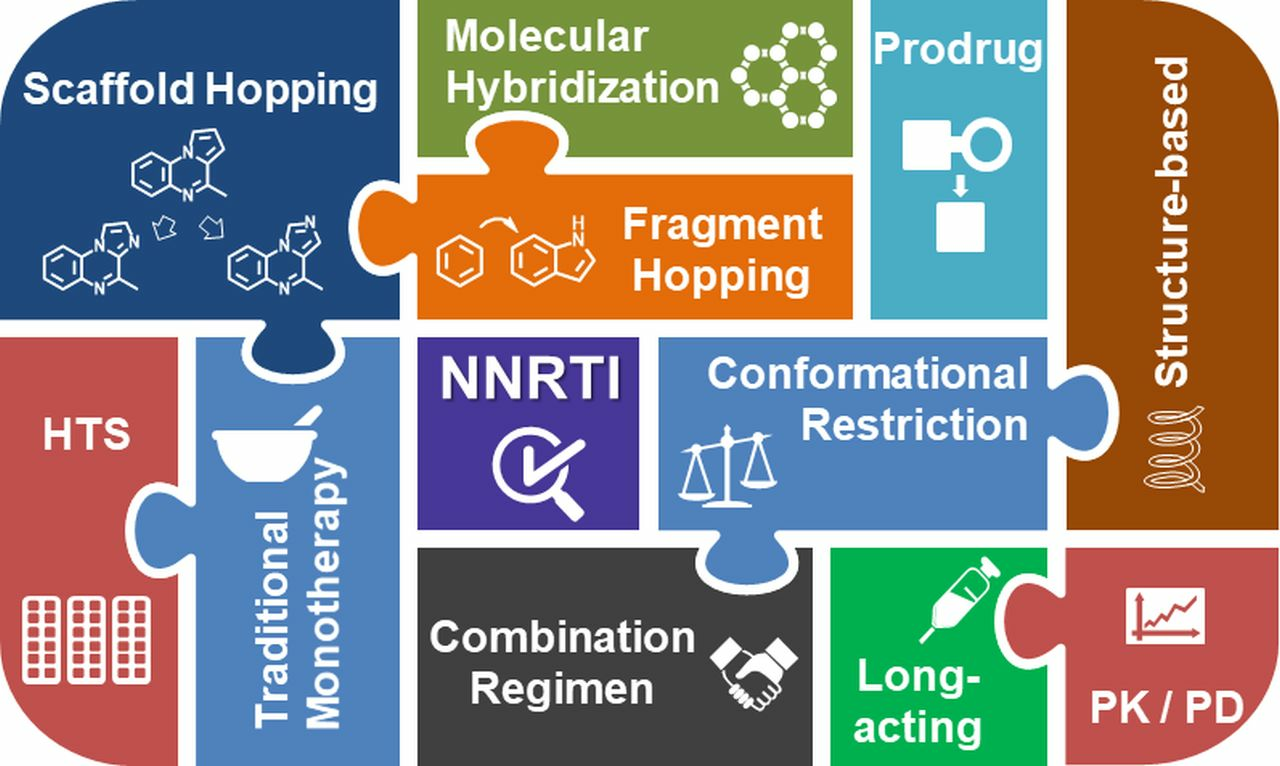
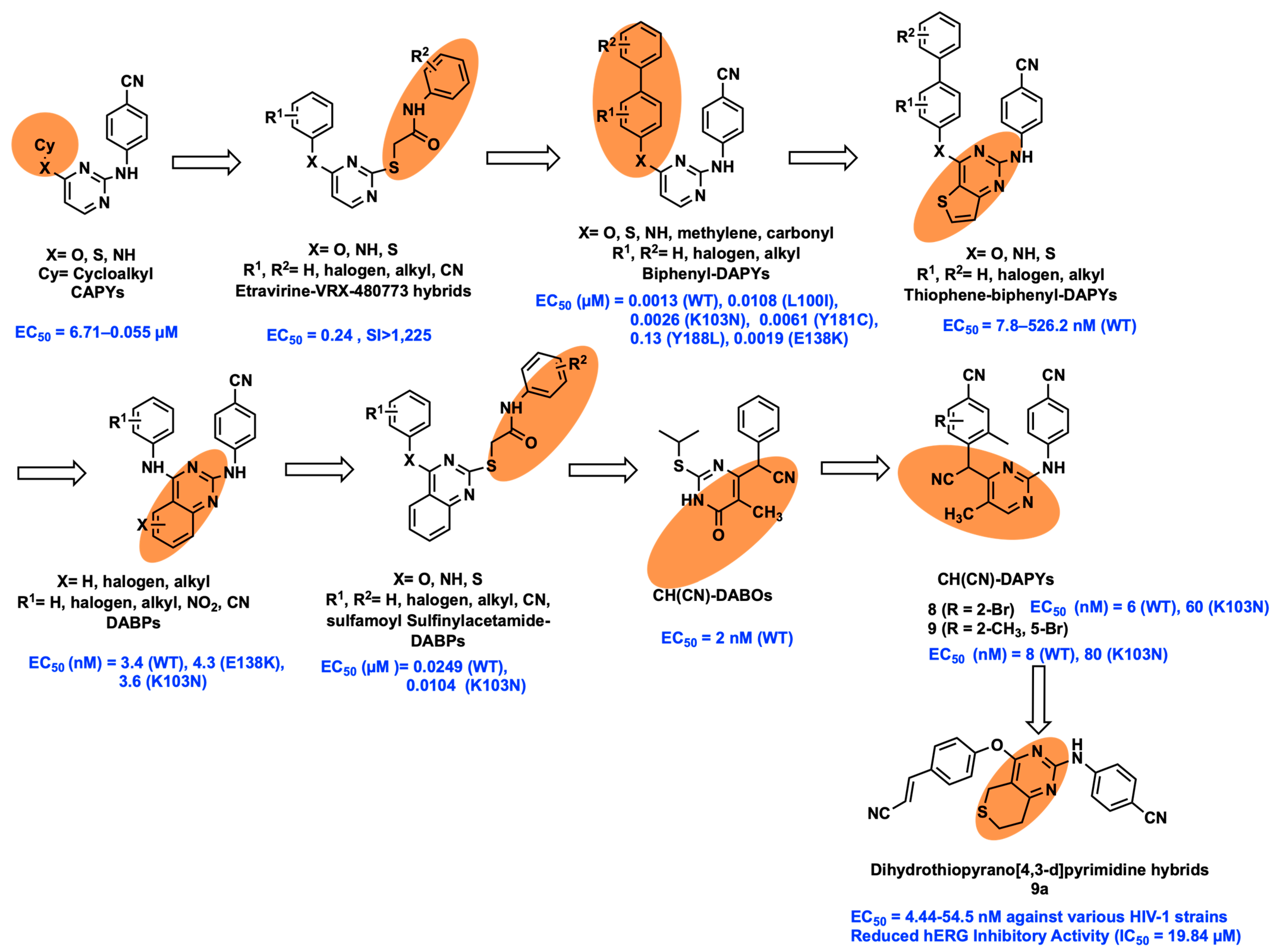
2.1. Molecular Hybridization
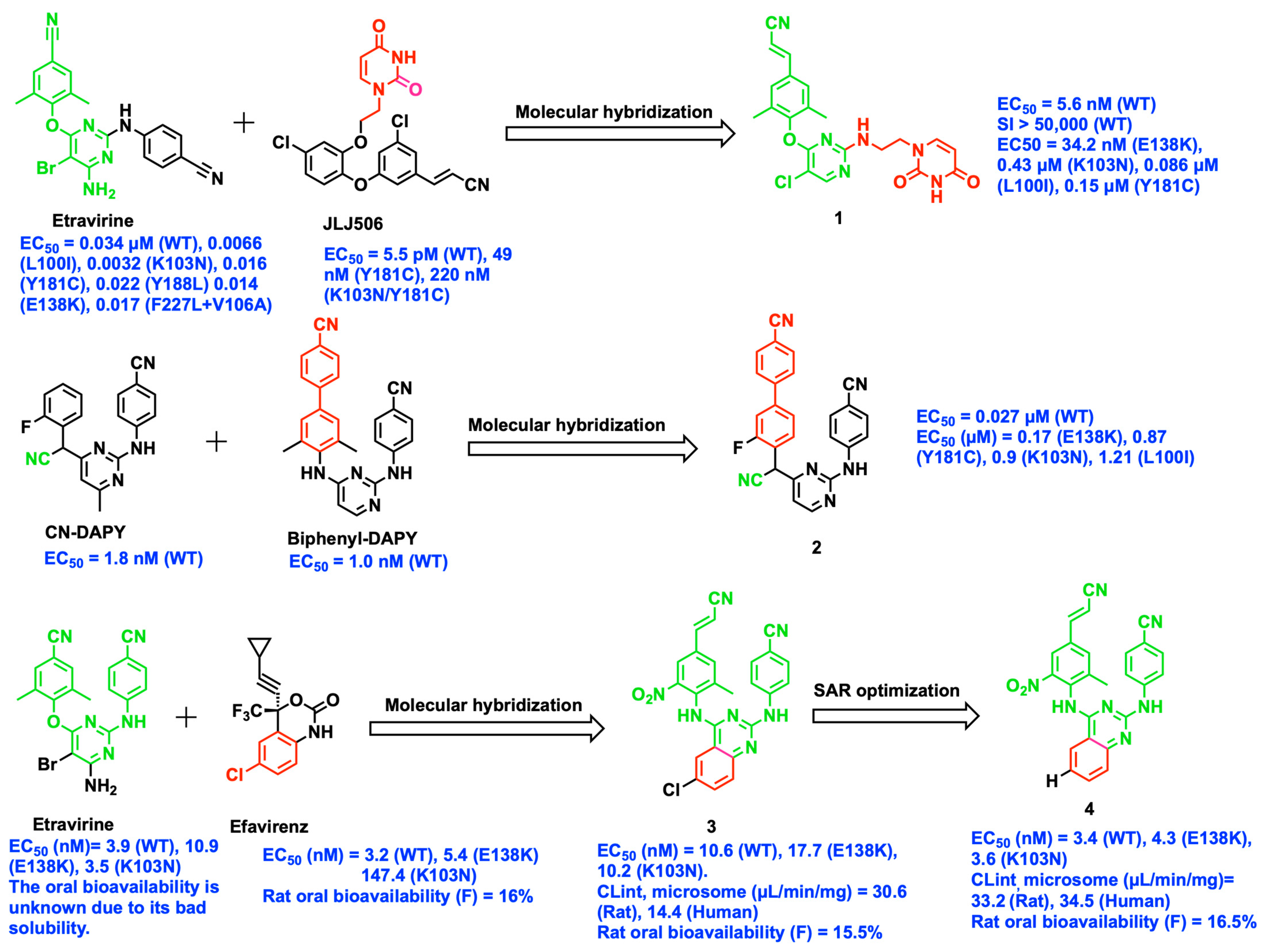
2.2. Bioisosteric Replacement
2.3. Scaffold Hopping
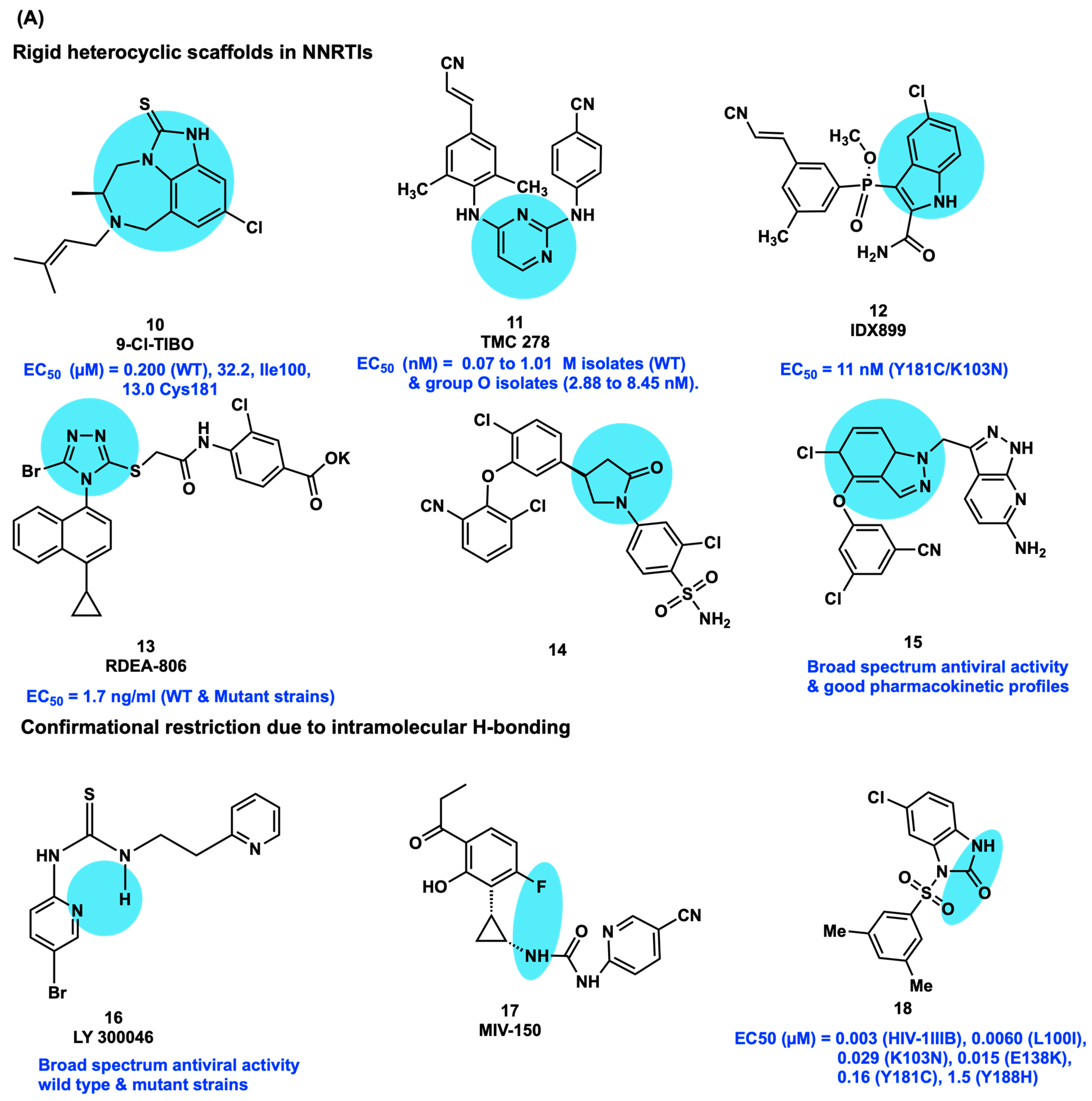
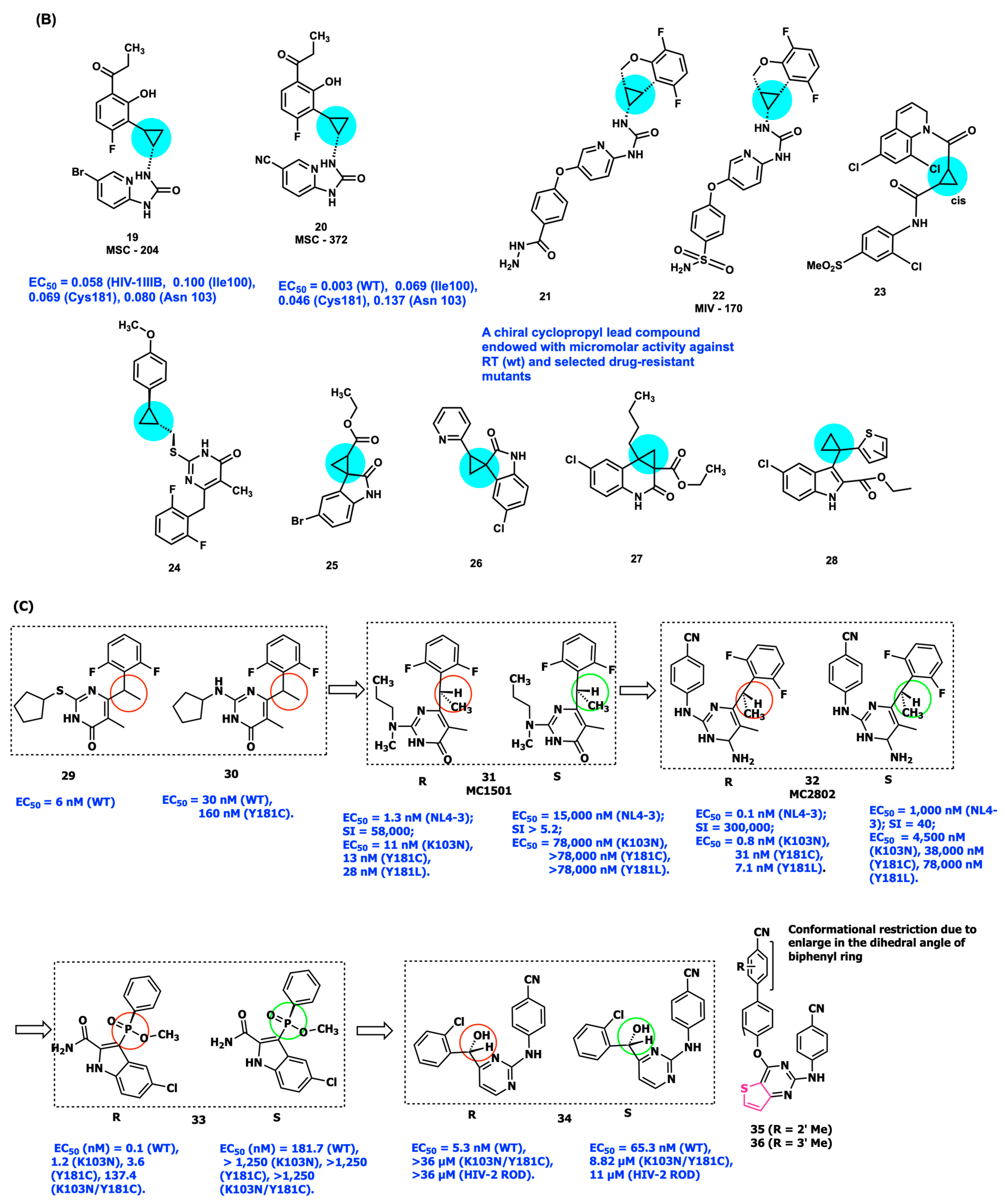
2.4. Conformational Restriction
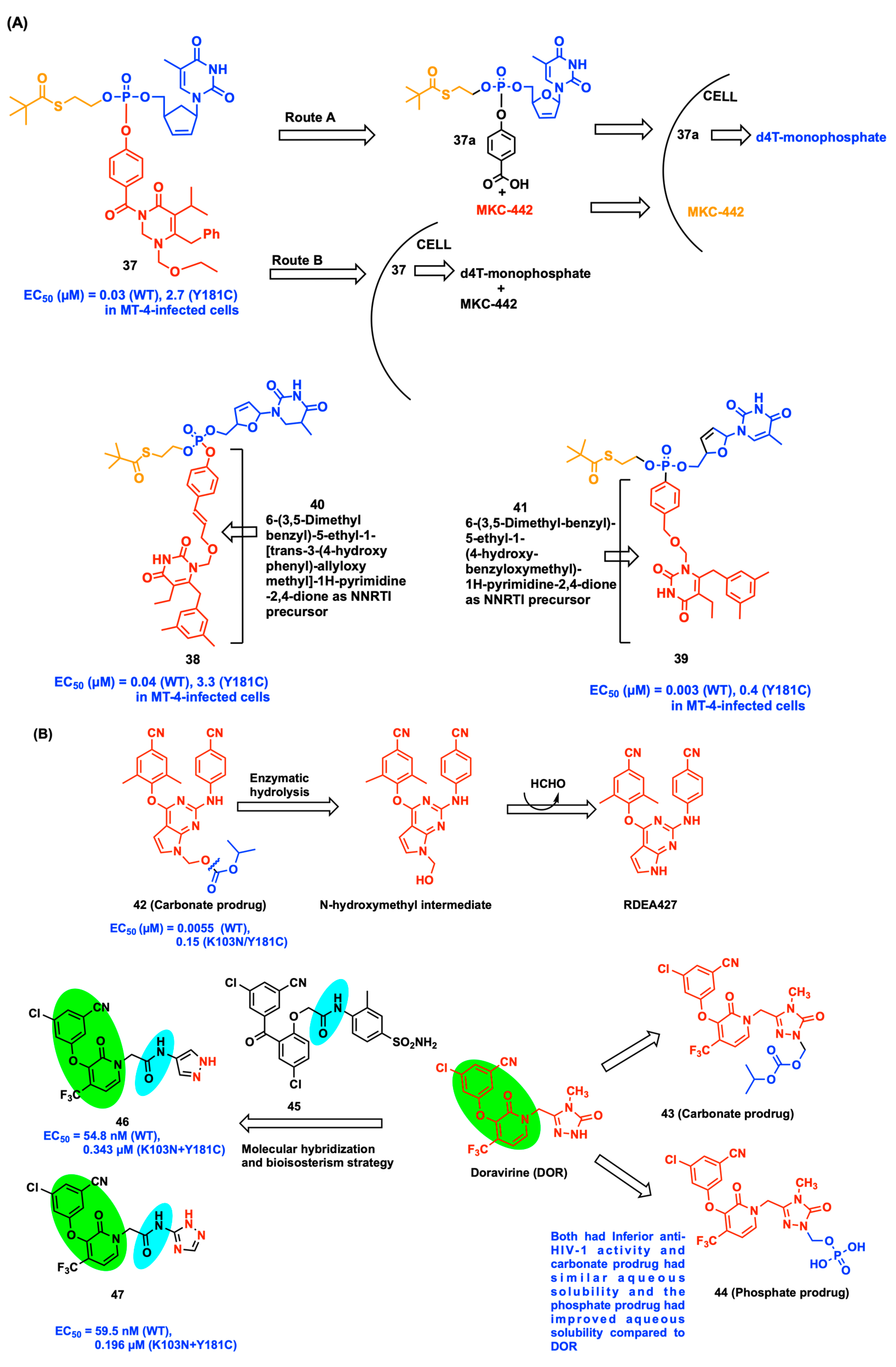
2.5. Prodrug
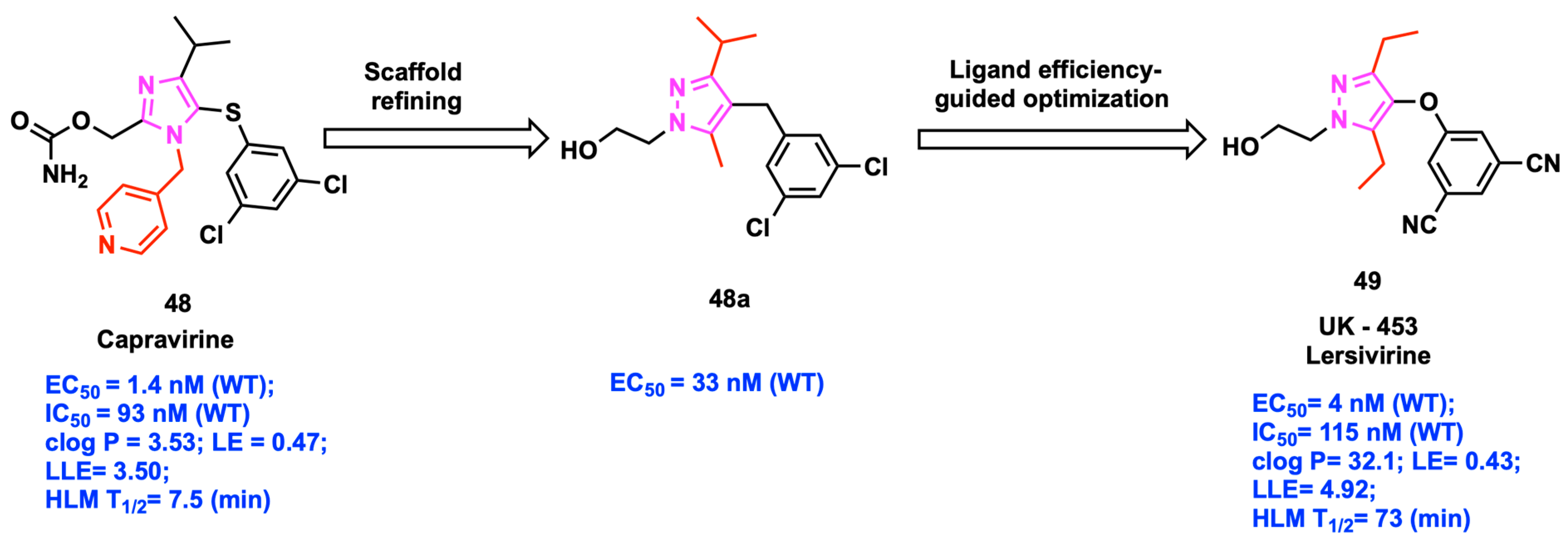
2.6. Ligand-Lipophilicity Efficiency (LLE)
2.7. Structure-Based Optimization
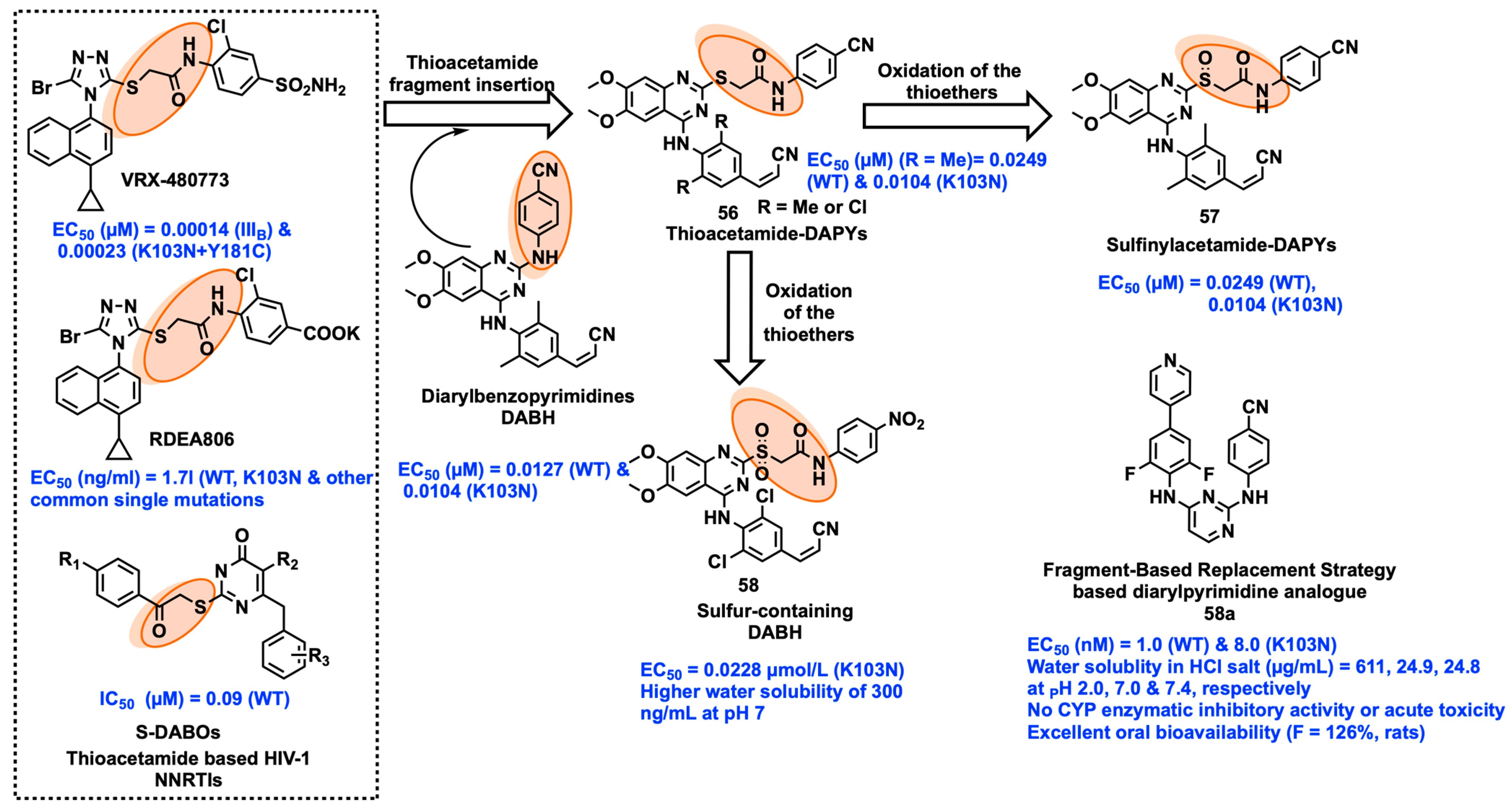
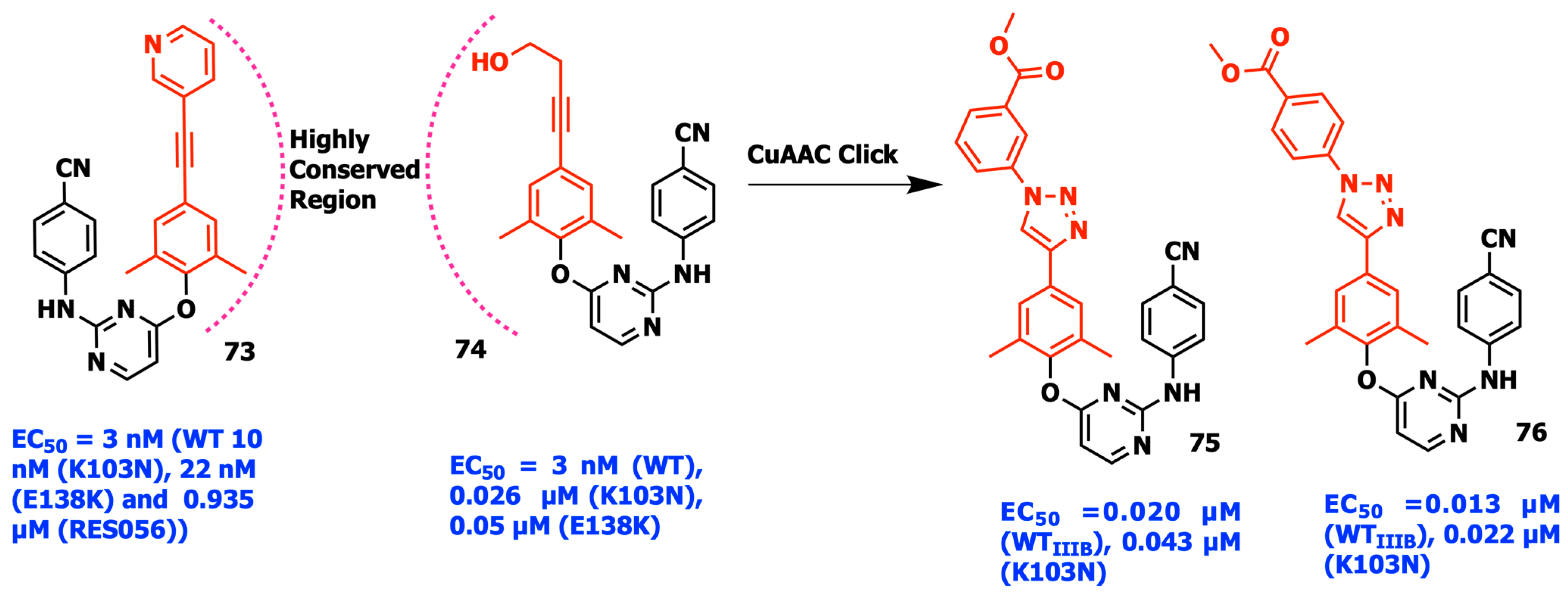
2.8. Fragment Hopping
2.9. High-Throughput Screening
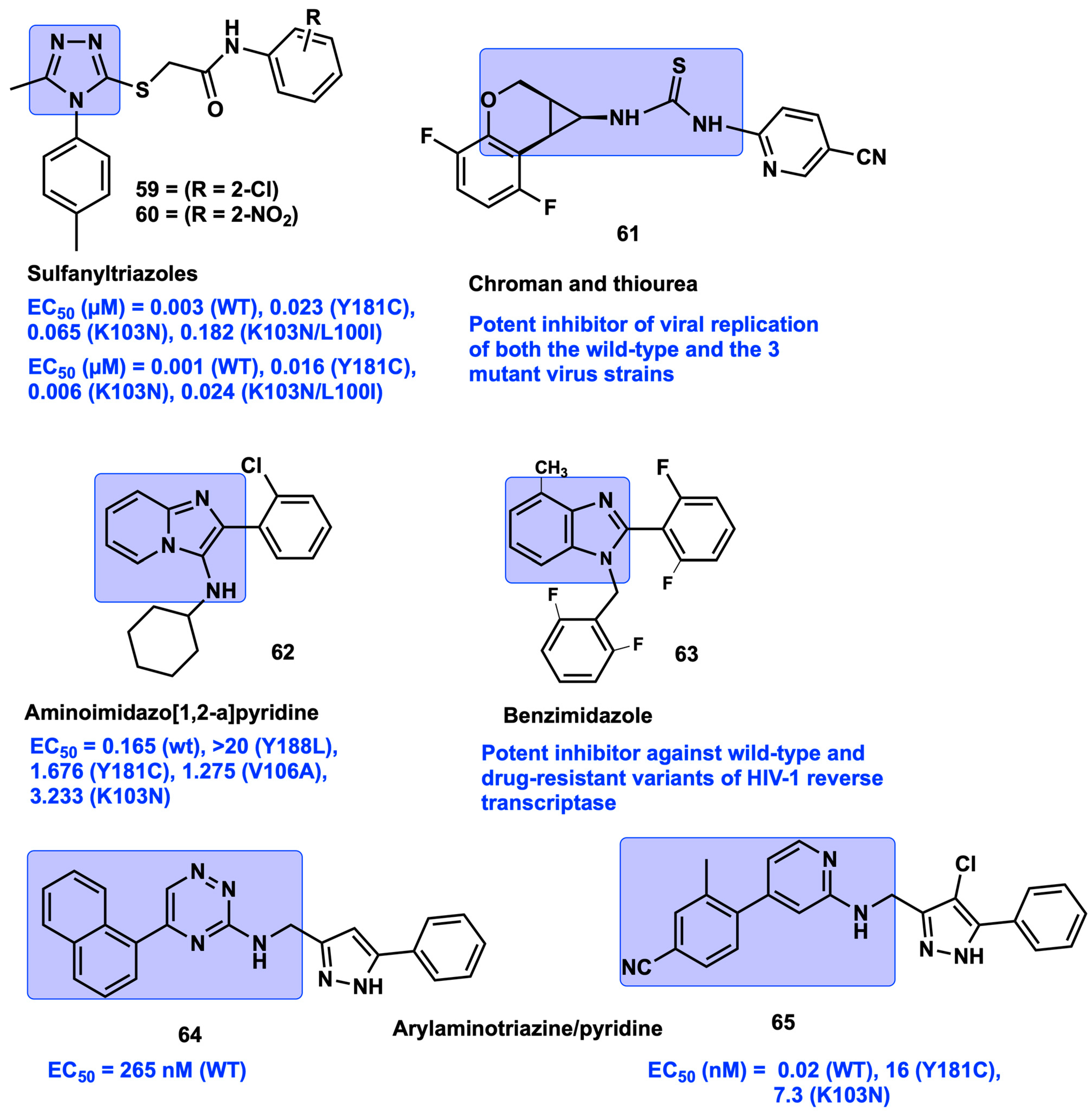
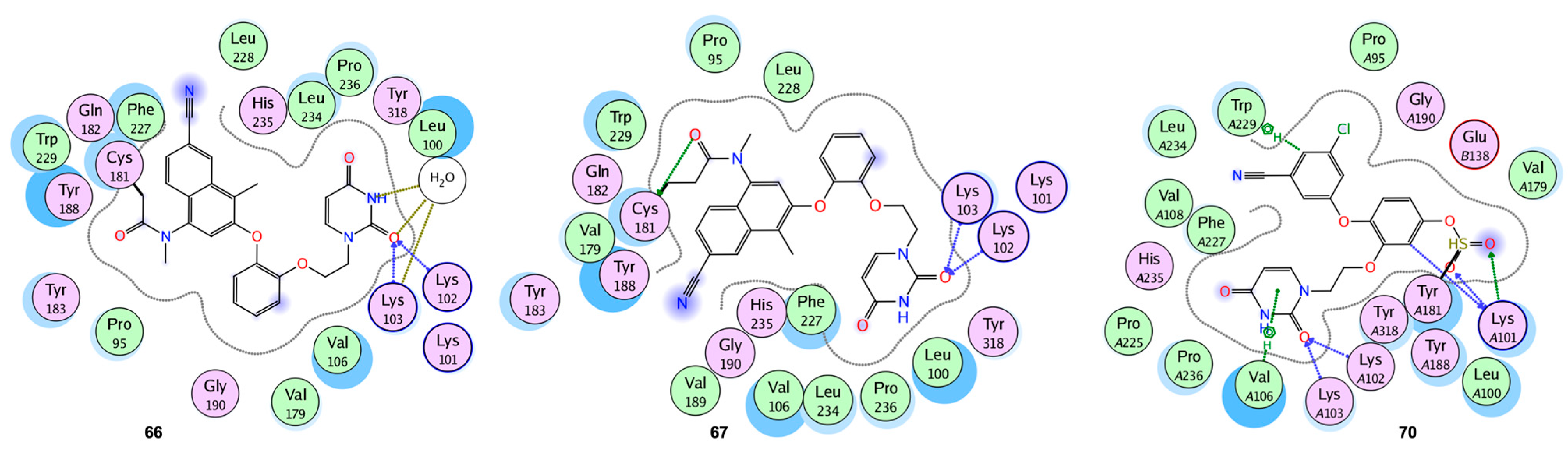
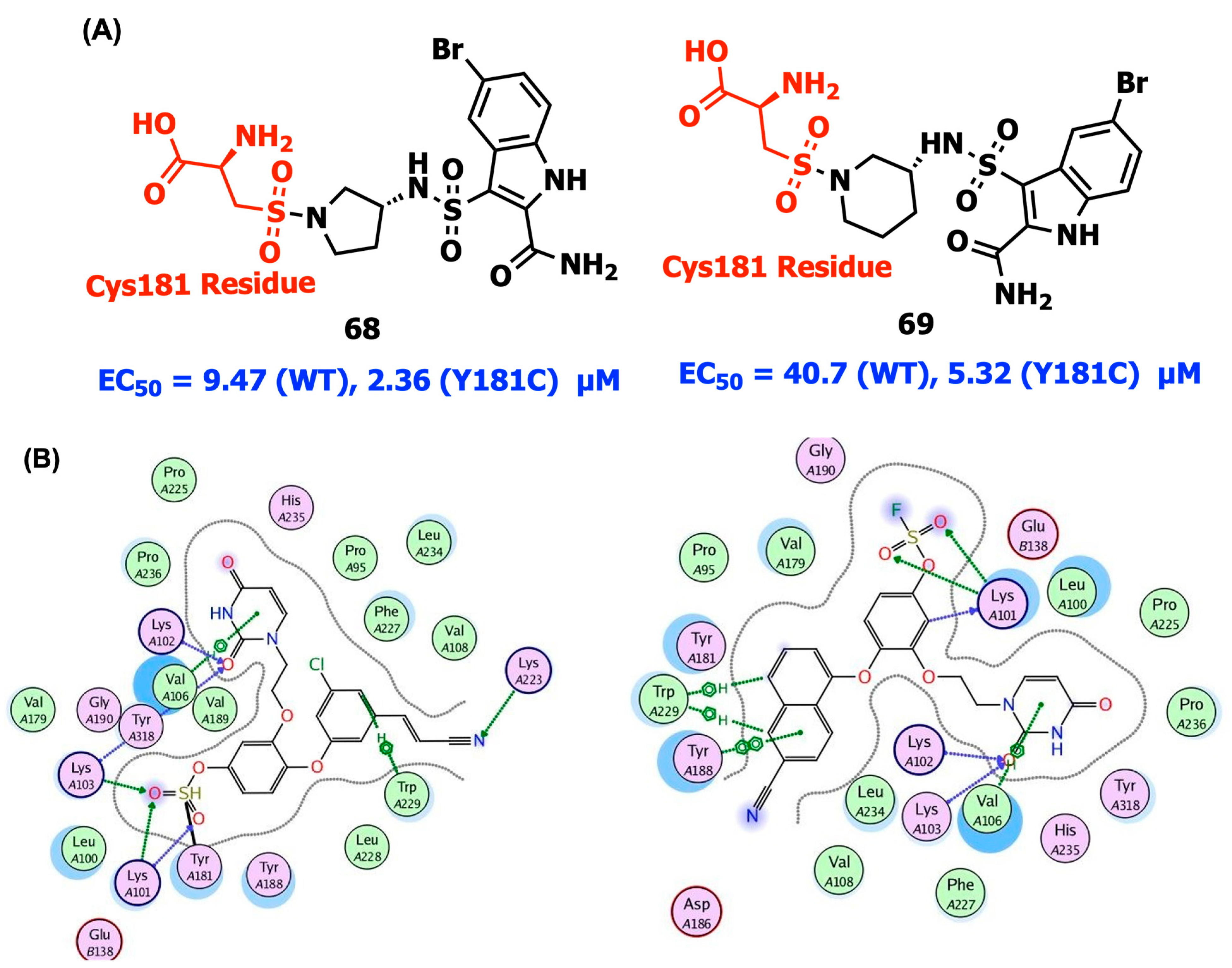
2.10. Covalent Bonding Inhibition
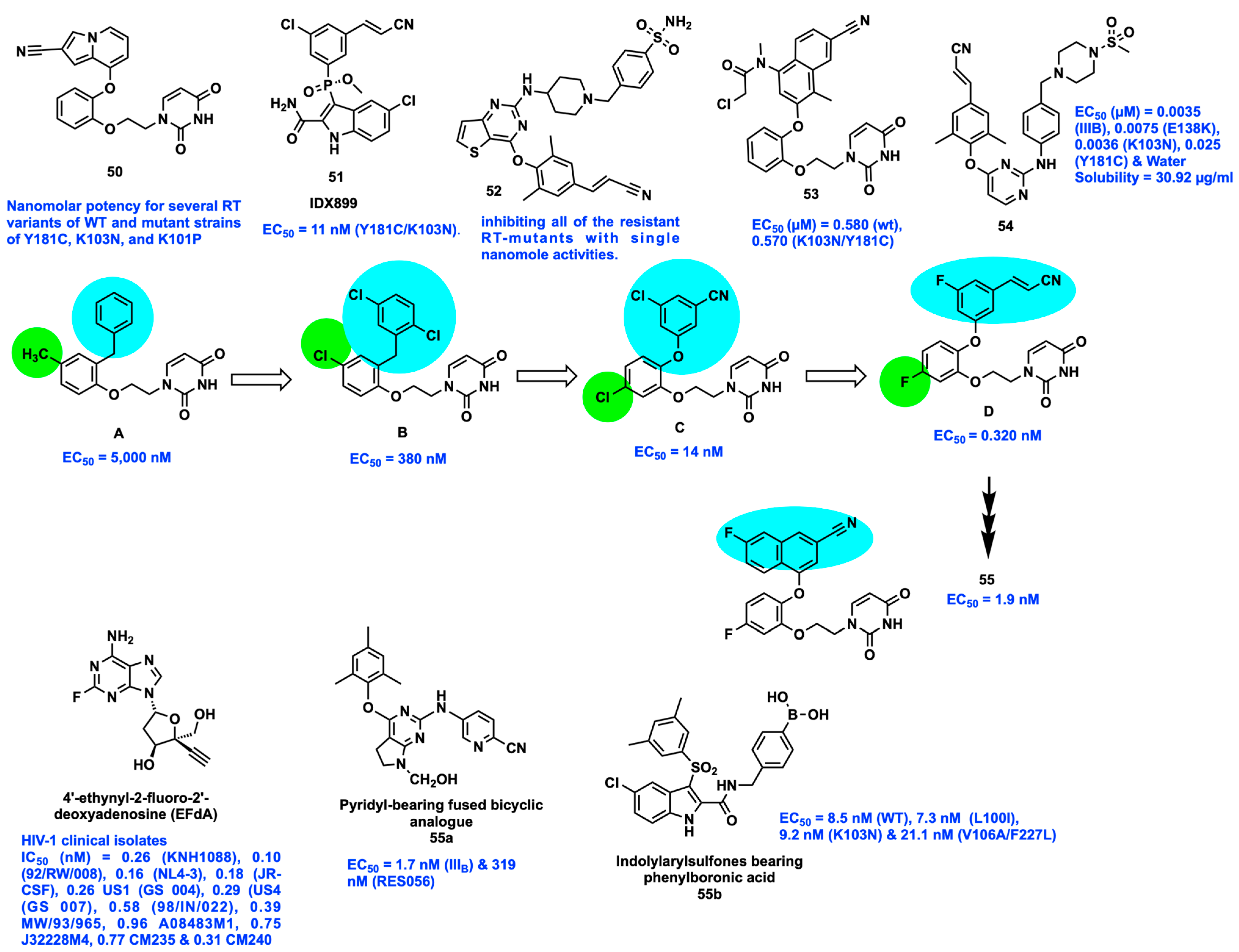
2.11. Targeting Highly Hydrophobic Channels
2.12. Targeting the Hydrophilic Solvent-Exposed Region
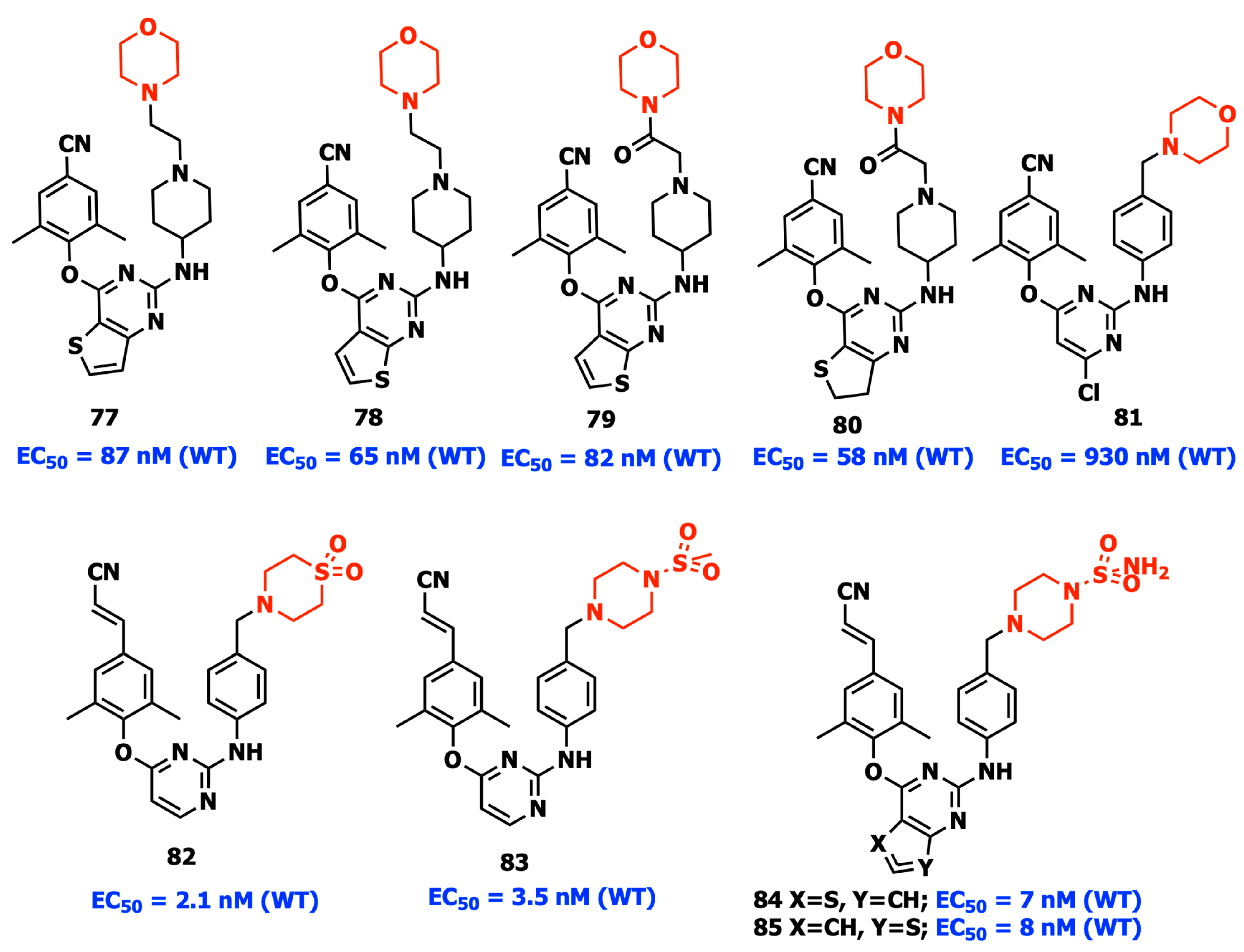
2.13. Targeting Dual Sites
2.14. Traditional Monotherapy
2.15. Combination Regimen
2.16. Formulation
3. Comparative Assessment of the Strategies in the Development of Novel NNRTIs
4. Conclusions
5. Perspective
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pierson, T.; McArthur, J.; Siliciano, R.F. Reservoirs for HIV-1: Mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu. Rev. Immunol. 2000, 18, 665–708. [Google Scholar] [CrossRef]
- Zhan, P.; Pannecouque, C.; De Clercq, E.; Liu, X. Anti-HIV Drug Discovery and Development: Current Innovations and Future Trends. J. Med. Chem. 2016, 59, 2849–2878. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Rong, L.; Liang, C.; Wainberg, M.A. Reverse Transcriptase Inhibitors Can Selectively Block the Synthesis of Differently Sized Viral DNA Transcripts in Cells Acutely Infected with Human Immunodeficiency Virus Type 1. J. Virol. 1999, 73, 6700–6707. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; De Clercq, E.; Li, G. Current and emerging non-nucleoside reverse transcriptase inhibitors (NNRTIs) for HIV-1 treatment. Expert Opin. Drug Metab. Toxicol. 2019, 15, 813–829. [Google Scholar] [CrossRef]
- Vanangamudi, M.; Poongavanam, V.; Namasivayam, V. HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors: SAR and Lead Optimization Using CoMFA and CoMSIA Studies (1995–2016). Curr. Med. Chem. 2017, 24, 3774–3812. [Google Scholar] [CrossRef]
- Namasivayam, V.; Vanangamudi, M.; Kramer, V.G.; Kurup, S.; Zhan, P.; Liu, X.; Kongsted, J.; Byrareddy, S.N. The Journey of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) from Lab to Clinic. J. Med. Chem. 2019, 62, 4851–4883. [Google Scholar] [CrossRef]
- Battini, L.; Bollini, M. Challenges and approaches in the discovery of human immunodeficiency virus type-1 non-nucleoside reverse transcriptase inhibitors. Med. Res. Rev. 2019, 39, 1235–1273. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Pannecouque, C.; De Clercq, E.; Chen, F. Development of non-nucleoside reverse transcriptase inhibitors (NNRTIs): Our past twenty years. Acta Pharm. Sin. B 2020, 10, 961–978. [Google Scholar] [CrossRef]
- Grande, F.; Ioele, G.; Occhiuzzi, M.A.; De Luca, M.; Mazzotta, E.; Ragno, G.; Garofalo, A.; Muzzalupo, R. Reverse Transcriptase Inhibitors Nanosystems Designed for Drug Stability and Controlled Delivery. Pharmaceutics 2019, 11, 197. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, J.; Gao, P.; Sun, L.; Song, Y.; Kang, D.; Liu, X.; Zhan, P. Efficient drug discovery by rational lead hybridization based on crystallographic overlay. Drug Discov. Today 2019, 24, 805–813. [Google Scholar] [CrossRef]
- Zhang, H.; Tian, Y.; Kang, D.; Huo, Z.; Zhou, Z.; Liu, H.; De Clercq, E.; Pannecouque, C.; Zhan, P.; Liu, X. Discovery of uracil-bearing DAPYs derivatives as novel HIV-1 NNRTIs via crystallographic overlay-based molecular hybridization. Eur. J. Med. Chem. 2017, 130, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Han, S.; Yang, Y.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F.E. Design of Biphenyl-Substituted Diarylpyrimidines with a Cyanomethyl Linker as HIV-1 NNRTIs via a Molecular Hybridization Strategy. Molecules 2020, 25, 1050. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Sang, Y.; Wu, Y.; Tao, Y.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F.E. Molecular Hybridization-Inspired Optimization of Diarylbenzopyrimidines as HIV-1 Nonnucleoside Reverse Transcriptase Inhibitors with Improved Activity against K103N and E138K Mutants and Pharmacokinetic Profiles. ACS Infect. Dis. 2020, 6, 787–801. [Google Scholar] [CrossRef]
- Ding, L.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F.E. Discovery of Novel Pyridine-Dimethyl-Phenyl-DAPY Hybrids by Molecular Fusing of Methyl-Pyrimidine-DAPYs and Difluoro-Pyridinyl-DAPYs: Improving the Druggability toward High Inhibitory Activity, Solubility, Safety, and PK. J. Med. Chem. 2022, 65, 2122–2138. [Google Scholar] [CrossRef]
- Lima, L.M.; Barreiro, E.J. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Dick, A.; Cocklin, S. Bioisosteric Replacement as a Tool in Anti-HIV Drug Design. Pharmaceuticals 2020, 13, 36. [Google Scholar] [CrossRef]
- Zhan, P.; Li, X.; Li, Z.; Chen, X.; Tian, Y.; Chen, W.; Liu, X.; Pannecouque, C.; De Clercq, E. Structure-based bioisosterism design, synthesis and biological evaluation of novel 1,2,4-triazin-6-ylthioacetamides as potent HIV-1 NNRTIs. Bioorg. Med. Chem. Lett. 2012, 22, 7155–7162. [Google Scholar] [CrossRef]
- Kang, D.; Feng, D.; Sun, Y.; Fang, Z.; Wei, F.; De Clercq, E.; Pannecouque, C.; Liu, X.; Zhan, P. Structure-Based Bioisosterism Yields HIV-1 NNRTIs with Improved Drug-Resistance Profiles and Favorable Pharmacokinetic Properties. J. Med. Chem. 2020, 63, 4837–4848. [Google Scholar] [CrossRef]
- Chen, X.; Ding, L.; Tao, Y.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F.E. Bioisosterism-based design and enantiomeric profiling of chiral hydroxyl-substituted biphenyl-diarylpyrimidine nonnucleoside HIV-1 reverse transcriptase inhibitors. Eur. J. Med. Chem. 2020, 202, 112549. [Google Scholar] [CrossRef]
- Böhm, H.J.; Flohr, A.; Stahl, M. Scaffold hopping. Drug Discov. Today Technol. 2004, 1, 217–224. [Google Scholar] [CrossRef]
- Hu, Y.; Stumpfe, D.; Bajorath, J. Recent Advances in Scaffold Hopping. J. Med. Chem. 2017, 60, 1238–1246. [Google Scholar] [CrossRef]
- Zhao, H. Scaffold selection and scaffold hopping in lead generation: A medicinal chemistry perspective. Drug Discov. Today 2007, 12, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Han, S.; Han, S.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F. Follow on-based optimization of the biphenyl-DAPYs as HIV-1 nonnucleoside reverse transcriptase inhibitors against the wild-type and mutant strains. Bioorg. Chem. 2019, 89, 102974. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Han, S.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F. Ligand-Based Design of Nondimethylphenyl-Diarylpyrimidines with Improved Metabolic Stability, Safety, and Oral Pharmacokinetic Profiles. J. Med. Chem. 2019, 62, 11430–11436. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Yang, S.Q.; He, Q.Q.; Ma, X.D.; Chen, F.E.; Dai, H.F.; Clercq, E.D.; Balzarini, J.; Pannecouque, C. Design, synthesis and biological evaluation of cycloalkyl arylpyrimidines (CAPYs) as HIV-1 NNRTIs. Bioorg. Med. Chem. 2011, 19, 7093–7099. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Yin, H.; De Clercq, E.; Pannecouque, C.; Meng, G.; Chen, F. Discovery of biphenyl-substituted diarylpyrimidines as non-nucleoside reverse transcriptase inhibitors with high potency against wild-type and mutant HIV-1. Eur. J. Med. Chem. 2018, 145, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Han, S.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F. Conformational restriction design of thiophene-biphenyl-DAPY HIV-1 non-nucleoside reverse transcriptase inhibitors. Eur. J. Med. Chem. 2019, 182, 111603. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.Y.; Tao, Y.; Wang, Y.F.; Mao, T.Q.; Yin, H.; Chen, F.E.; Piao, H.R.; De Clercq, E.; Daelemans, D.; Pannecouque, C. Hybrid chemistry. Part 4: Discovery of etravirine-VRX-480773 hybrids as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. 2015, 23, 4248–4255. [Google Scholar] [CrossRef]
- Han, S.; Sang, Y.; Wu, Y.; Tao, Y.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F.E. Fragment hopping-based discovery of novel sulfinylacetamide-diarylpyrimidines (DAPYs) as HIV-1 nonnucleoside reverse transcriptase inhibitors. Eur. J. Med. Chem. 2020, 185, 111874. [Google Scholar] [CrossRef]
- Li, T.T.; Pannecouque, C.; De Clercq, E.; Zhuang, C.L.; Chen, F.E. Scaffold Hopping in Discovery of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors: From CH(CN)-DABOs to CH(CN)-DAPYs. Molecules 2020, 25, 1581. [Google Scholar] [CrossRef]
- Wang, Z.; Zalloum, W.A.; Wang, W.; Jiang, X.; De Clercq, E.; Pannecouque, C.; Kang, D.; Zhan, P.; Liu, X. Discovery of Novel Dihydrothiopyrano[4,3-d]pyrimidine Derivatives as Potent HIV-1 NNRTIs with Significantly Reduced hERG Inhibitory Activity and Improved Resistance Profiles. J. Med. Chem. 2021, 64, 13658–13675. [Google Scholar] [CrossRef]
- Fang, Z.; Song, Y.; Zhan, P.; Zhang, Q.; Liu, X. Conformational restriction: An effective tactic in ‘follow-on’-based drug discovery. Future Med. Chem. 2014, 6, 885–901. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, R.; Andries, K.; Desmyter, J.; Schols, D.; Kukla, M.J.; Breslin, H.J.; Raeymaeckers, A.; Van Gelder, J.; Woestenborghs, R.; Heykants, J.; et al. Potent and selective inhibition of HIV-1 replication in vitro by a novel series of TIBO derivatives. Nature 1990, 343, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Schafer, J.J.; Short, W.R. Rilpivirine, a novel non-nucleoside reverse transcriptase inhibitor for the management of HIV-1 infection: A systematic review. Antivir. Ther. 2012, 17, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, O.M.; Kaczor, R.L. IDX-899, an aryl phosphinate-indole non-nucleoside reverse transcriptase inhibitor for the potential treatment of HIV infection. Curr. Opin. Investig. Drugs 2010, 11, 237–245. [Google Scholar]
- Moyle, G.; Boffito, M.; Stoehr, A.; Rieger, A.; Shen, Z.; Manhard, K.; Sheedy, B.; Hingorani, V.; Raney, A.; Nguyen, M.; et al. Phase 2a randomized controlled trial of short-term activity, safety, and pharmacokinetics of a novel nonnucleoside reverse transcriptase inhibitor, RDEA806, in HIV-1-positive, antiretroviral-naive subjects. Antimicrob. Agents Chemother. 2010, 54, 3170–3178. [Google Scholar] [CrossRef]
- Gomez, R.; Jolly, S.J.; Williams, T.; Vacca, J.P.; Torrent, M.; McGaughey, G.; Lai, M.T.; Felock, P.; Munshi, V.; Distefano, D.; et al. Design and synthesis of conformationally constrained inhibitors of non-nucleoside reverse transcriptase. J. Med. Chem. 2011, 54, 7920–7933. [Google Scholar] [CrossRef]
- Cantrell, A.S.; Engelhardt, P.; Högberg, M.; Jaskunas, S.R.; Johansson, N.G.; Jordan, C.L.; Kangasmetsä, J.; Kinnick, M.D.; Lind, P.; Morin, J.M., Jr.; et al. Phenethylthiazolylthiourea (PETT) compounds as a new class of HIV-1 reverse transcriptase inhibitors. 2. Synthesis and further structure-activity relationship studies of PETT analogs. J. Med. Chem. 1996, 39, 4261–4274. [Google Scholar] [CrossRef]
- Barreca, M.L.; Rao, A.; De Luca, L.; Iraci, N.; Monforte, A.M.; Maga, G.; De Clercq, E.; Pannecouque, C.; Balzarini, J.; Chimirri, A. Discovery of novel benzimidazolones as potent non-nucleoside reverse transcriptase inhibitors active against wild-type and mutant HIV-1 strains. Bioorg. Med. Chem. Lett. 2007, 17, 1956–1960. [Google Scholar] [CrossRef]
- Ren, J.; Diprose, J.; Warren, J.; Esnouf, R.M.; Bird, L.E.; Ikemizu, S.; Slater, M.; Milton, J.; Balzarini, J.; Stuart, D.I.; et al. Phenylethylthiazolylthiourea (PETT) non-nucleoside inhibitors of HIV-1 and HIV-2 reverse transcriptases. Structural and biochemical analyses. J. Biol. Chem. 2000, 275, 5633–5639. [Google Scholar] [CrossRef]
- Hogberg, M.; Sahlberg, C.; Engelhardt, P.; Noreen, R.; Kangasmetsa, J.; Johansson, N.G.; Oberg, B.; Vrang, L.; Zhang, H.; Sahlberg, B.L.; et al. Urea-PETT compounds as a new class of HIV-1 reverse transcriptase inhibitors. 3. Synthesis and further structure-activity relationship studies of PETT analogues. J. Med. Chem. 2000, 43, 304. [Google Scholar] [CrossRef] [PubMed]
- Su, D.S.; Lim, J.J.; Tinney, E.; Wan, B.L.; Young, M.B.; Anderson, K.D.; Rudd, D.; Munshi, V.; Bahnck, C.; Felock, P.J.; et al. Substituted tetrahydroquinolines as potent allosteric inhibitors of reverse transcriptase and its key mutants. Bioorg. Med. Chem. Lett. 2009, 19, 5119–5123. [Google Scholar] [CrossRef] [PubMed]
- Radi, M.; Maga, G.; Alongi, M.; Angeli, L.; Samuele, A.; Zanoli, S.; Bellucci, L.; Tafi, A.; Casaluce, G.; Giorgi, G.; et al. Discovery of chiral cyclopropyl dihydro-alkylthio-benzyl-oxopyrimidine (S-DABO) derivatives as potent HIV-1 reverse transcriptase inhibitors with high activity against clinically relevant mutants. J. Med. Chem. 2009, 52, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Kuhen, K.L.; Wolff, K.; Yin, H.; Bieza, K.; Caldwell, J.; Bursulaya, B.; Wu, T.Y.; He, Y. Design, synthesis and biological evaluations of novel oxindoles as HIV-1 non-nucleoside reverse transcriptase inhibitors. Part I. Bioorg. Med. Chem. Lett. 2006, 16, 2105–2108. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Kuhen, K.L.; Wolff, K.; Yin, H.; Bieza, K.; Caldwell, J.; Bursulaya, B.; Tuntland, T.; Zhang, K.; Karanewsky, D.; et al. Design, synthesis, and biological evaluations of novel oxindoles as HIV-1 non-nucleoside reverse transcriptase inhibitors. Part 2. Bioorg. Med. Chem. Lett. 2006, 16, 2109–2112. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.; Kuhen, K.L.; Anaclerio, B.; Wu, B.; Wolff, K.; Yin, H.; Bursulaya, B.; Caldwell, J.; Karanewsky, D.; He, Y. Design, synthesis, and biological evaluations of novel quinolones as HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 4246–4251. [Google Scholar] [CrossRef]
- Hassam, M.; Basson, A.E.; Liotta, D.C.; Morris, L.; van Otterlo, W.A.; Pelly, S.C. Novel Cyclopropyl-Indole Derivatives as HIV Non-Nucleoside Reverse Transcriptase Inhibitors. ACS Med. Chem. Lett. 2012, 3, 470–475. [Google Scholar] [CrossRef]
- Rotili, D.; Samuele, A.; Tarantino, D.; Ragno, R.; Musmuca, I.; Ballante, F.; Botta, G.; Morera, L.; Pierini, M.; Cirilli, R.; et al. 2-(Alkyl/Aryl)Amino-6-Benzylpyrimidin-4(3H)-ones as Inhibitors of Wild-Type and Mutant HIV-1: Enantioselectivity Studies. J. Med. Chem. 2012, 55, 3558–3562. [Google Scholar] [CrossRef]
- Ragno, R.; Mai, A.; Sbardella, G.; Artico, M.; Massa, S.; Musiu, C.; Mura, M.; Marturana, F.; Cadeddu, A.; La Colla, P. Computer-aided design, synthesis, and anti-HIV-1 activity in vitro of 2-alkylamino-6-[1-(2,6-difluorophenyl)alkyl]-3,4-dihydro-5-alkylpyrimidin-4(3H)-ones as novel potent non-nucleoside reverse transcriptase inhibitors, also active against the Y181C variant. J. Med. Chem. 2004, 47, 928–934. [Google Scholar] [CrossRef]
- Alexandre, F.R.; Amador, A.; Bot, S.; Caillet, C.; Convard, T.; Jakubik, J.; Musiu, C.; Poddesu, B.; Vargiu, L.; Liuzzi, M.; et al. Synthesis and biological evaluation of aryl-phospho-indole as novel HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Med. Chem. 2011, 54, 392–395. [Google Scholar] [CrossRef]
- Gu, S.X.; Li, Z.M.; Ma, X.D.; Yang, S.Q.; He, Q.Q.; Chen, F.E.; De Clercq, E.; Balzarini, J.; Pannecouque, C. Chiral resolution, absolute configuration assignment and biological activity of racemic diarylpyrimidine CH(OH)-DAPY as potent nonnucleoside HIV-1 reverse transcriptase inhibitors. Eur. J. Med. Chem. 2012, 53, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Calogeropoulou, T.; Detsi, A.; Lekkas, E.; Koufaki, M. Strategies in the design of prodrugs of anti-HIV agents. Curr. Top. Med. Chem. 2003, 3, 1467–1495. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y. Recent progress in prodrug design strategies based on generally applicable modifications. Bioorg. Med. Chem. Lett. 2017, 27, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.; Jørgensen, P.T.; Nielsen, C.; Hansen, T.H.; Nielsen, J.; Pedersen, E.B. Synthesis and evaluation of double-prodrugs against HIV. Conjugation of D4T with 6-benzyl-1-(ethoxymethyl)-5-isopropyluracil (MKC-442, emivirine)-type reverse transcriptase inhibitors via the SATE prodrug approach. J. Med. Chem. 2005, 48, 1211–1220. [Google Scholar] [CrossRef]
- Huang, B.; Liu, X.; Tian, Y.; Kang, D.; Zhou, Z.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; Zhan, P.; Liu, X. First discovery of a potential carbonate prodrug of NNRTI drug candidate RDEA427 with submicromolar inhibitory activity against HIV-1 K103N/Y181C double mutant strain. Bioorg. Med. Chem. Lett. 2018, 28, 1348–1351. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yu, Z.; Kang, D.; Zhang, J.; Tian, Y.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; Zhan, P.; Liu, X. Design, synthesis and biological evaluation of novel acetamide-substituted doravirine and its prodrugs as potent HIV-1 NNRTIs. Bioorg. Med. Chem. 2019, 27, 447–456. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Abad-Zapatero, C.; Perišić, O.; Wass, J.; Bento, A.P.; Overington, J.; Al-Lazikani, B.; Johnson, M.E. Ligand efficiency indices for an effective mapping of chemico-biological space: The concept of an atlas-like representation. Drug Discov. Today 2010, 15, 804–811. [Google Scholar] [CrossRef]
- Mowbray, C.E.; Burt, C.; Corbau, R.; Perros, M.; Tran, I.; Stupple, P.A.; Webster, R.; Wood, A. Pyrazole NNRTIs 1: Design and initial optimisation of a novel template. Bioorg. Med. Chem. Lett. 2009, 19, 5599–5602. [Google Scholar] [CrossRef]
- Mowbray, C.E.; Corbau, R.; Hawes, M.; Jones, L.H.; Mills, J.E.; Perros, M.; Selby, M.D.; Stupple, P.A.; Webster, R.; Wood, A. Pyrazole NNRTIs 3: Optimisation of physicochemical properties. Bioorg. Med. Chem. Lett. 2009, 19, 5603–5606. [Google Scholar] [CrossRef]
- Mowbray, C.E.; Burt, C.; Corbau, R.; Gayton, S.; Hawes, M.; Perros, M.; Tran, I.; Price, D.A.; Quinton, F.J.; Selby, M.D.; et al. Pyrazole NNRTIs 4: Selection of UK-453,061 (lersivirine) as a development candidate. Bioorg. Med. Chem. Lett. 2009, 19, 5857–5860. [Google Scholar] [CrossRef] [PubMed]
- Corbau, R.; Mori, J.; Phillips, C.; Fishburn, L.; Martin, A.; Mowbray, C.; Panton, W.; Smith-Burchnell, C.; Thornberry, A.; Ringrose, H.; et al. Lersivirine, a nonnucleoside reverse transcriptase inhibitor with activity against drug-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2010, 54, 4451–4463. [Google Scholar] [CrossRef] [PubMed]
- Fätkenheuer, G.; Staszewski, S.; Plettenburg, A.; Hackman, F.; Layton, G.; McFadyen, L.; Davis, J.; Jenkins, T.M. Activity, pharmacokinetics and safety of lersivirine (UK-453,061), a next-generation nonnucleoside reverse transcriptase inhibitor, during 7-day monotherapy in HIV-1-infected patients. AIDS 2009, 23, 2115–2122. [Google Scholar] [CrossRef]
- Platten, M.; Fätkenheuer, G. Lersivirine—A new drug for HIV infection therapy. Expert Opin. Investig. Drugs 2013, 22, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- García-Sosa, A.T.; Sild, S.; Takkis, K.; Maran, U. Combined approach using ligand efficiency, cross-docking, and antitarget hits for wild-type and drug-resistant Y181C HIV-1 reverse transcriptase. J. Chem. Inf. Model. 2011, 51, 2595–2611. [Google Scholar] [CrossRef] [PubMed]
- Bustanji, Y.; Al-Masri, I.M.; Qasem, A.; Al-Bakri, A.G.; Taha, M.O. In silico screening for non-nucleoside HIV-1 reverse transcriptase inhibitors using physicochemical filters and high-throughput docking followed by in vitro evaluation. Chem. Biol. Drug Des. 2009, 74, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Vanangamudi, M.; Nair, P.C.; Engels, S.E.M.; Palaniappan, S.; Namasivayam, V. Structural Insights to Human Immunodeficiency Virus (HIV-1) Targets and their Inhibition. In Antiviral Drug Discovery and Development; Liu, X., Zhan, P., Menéndez-Arias, L., Poongavanam, V., Eds.; Springer: Singapore, 2021. [Google Scholar] [CrossRef]
- Frey, K.M.; Tabassum, T. Current structure-based methods for designing non-nucleoside reverse transcriptase inhibitors. Future Med. 2019, 14, 537–544. [Google Scholar] [CrossRef]
- Bollini, M.; Domaoal, R.A.; Thakur, V.V.; Gallardo-Macias, R.; Spasov, K.A.; Anderson, K.S.; Jorgensen, W.L. Computationally-guided optimization of a docking hit to yield catechol diethers as potent anti-HIV agents. J. Med. Chem. 2011, 54, 8582–8591. [Google Scholar] [CrossRef]
- Frey, K.M.; Bollini, M.; Mislak, A.C.; Cisneros, J.A.; Gallardo-Macias, R.; Jorgensen, W.L.; Anderson, K.S. Crystal structures of HIV-1 reverse transcriptase with picomolar inhibitors reveal key interactions for drug design. J. Am. Chem. Soc. 2012, 134, 19501–19503. [Google Scholar] [CrossRef]
- Frey, K.M.; Puleo, D.E.; Spasov, K.A.; Bollini, M.; Jorgensen, W.L.; Anderson, K.S. Structure-based evaluation of non-nucleoside inhibitors with improved potency and solubility that target HIV reverse transcriptase variants. J. Med. Chem. 2015, 58, 2737–2745. [Google Scholar] [CrossRef]
- Dousson, C.; Alexandre, F.R.; Amador, A.; Bonaric, S.; Bot, S.; Caillet, C.; Convard, T.; da Costa, D.; Lioure, M.P.; Roland, A.; et al. Discovery of the Aryl-phospho-indole IDX899, a Highly Potent Anti-HIV Non-nucleoside Reverse Transcriptase Inhibitor. J. Med. Chem. 2016, 59, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.H.; Lee, W.G.; Spasov, K.A.; Cisneros, J.A.; Kudalkar, S.N.; Petrova, Z.O.; Buckingham, A.B.; Anderson, K.S.; Jorgensen, W.L. Covalent inhibitors for eradication of drug-resistant HIV-1 reverse transcriptase: From design to protein crystallography. Proc. Natl. Acad. Sci. USA 2017, 114, 9725–9730. [Google Scholar] [CrossRef]
- Huang, B.; Chen, W.; Zhao, T.; Li, Z.; Jiang, X.; Ginex, T.; Vílchez, D.; Luque, F.J.; Kang, D.; Gao, P.; et al. Exploiting the Tolerant Region I of the Non-Nucleoside Reverse Transcriptase Inhibitor (NNRTI) Binding Pocket: Discovery of Potent Diarylpyrimidine-Typed HIV-1 NNRTIs against Wild-Type and E138K Mutant Virus with Significantly Improved Water Solubility and Favorable Safety Profiles. J. Med. Chem. 2019, 62, 2083–2098. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Zhang, H.; Wang, Z.; Zhao, T.; Ginex, T.; Luque, F.J.; Yang, Y.; Wu, G.; Feng, D.; Wei, F.; et al. Identification of Dihydrofuro[3,4-d]pyrimidine Derivatives as Novel HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors with Promising Antiviral Activities and Desirable Physicochemical Properties. J. Med. Chem. 2019, 62, 1484–1501. [Google Scholar] [CrossRef]
- Kudalkar, S.N.; Beloor, J.; Quijano, E.; Spasov, K.A.; Lee, W.G.; Cisneros, J.A.; Saltzman, W.M.; Kumar, P.; Jorgensen, W.L.; Anderson, K.S. From in silico hit to long-acting late-stage preclinical candidate to combat HIV-1 infection. Proc. Natl. Acad. Sci. USA 2018, 115, E802–E811. [Google Scholar] [CrossRef]
- Huang, B.; Ginex, T.; Luque, F.J.; Jiang, X.; Gao, P.; Zhang, J.; Kang, D.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; et al. Structure-Based Design and Discovery of Pyridyl-Bearing Fused Bicyclic HIV-1 Inhibitors: Synthesis, Biological Characterization, and Molecular Modeling Studies. J. Med. Chem. 2021, 64, 13604–13621. [Google Scholar] [CrossRef]
- Xu, S.; Song, S.; Sun, L.; Gao, P.; Gao, S.; Ma, Y.; Kang, D.; Cheng, Y.; Zhang, X.; Cherukupalli, S.; et al. Indolylarylsulfones bearing phenylboronic acid and phenylboronate ester functionalities as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. 2022, 53, 116531. [Google Scholar] [CrossRef]
- Han, S.; Lei, Y.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F. Fragment-based discovery of sulfur-containing diarylbenzopyrimidines as novel nonnucleoside reverse transcriptase inhibitors. Chin. Chem. Lett. 2020, 31, 764–768. [Google Scholar] [CrossRef]
- Ding, L.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F.E. Improving Druggability of Novel Diarylpyrimidine NNRTIs by a Fragment-Based Replacement Strategy: From Biphenyl-DAPYs to Heteroaromatic-Biphenyl-DAPYs. J. Med. Chem. 2021, 64, 10297–10311. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, B.; Kuhen, K.L.; Bursulaya, B.; Nguyen, T.N.; Nguyen, D.G.; He, Y. Synthesis and biological evaluations of sulfanyltriazoles as novel HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 4174–4177. [Google Scholar] [CrossRef]
- Elinder, M.; Nordström, H.; Geitmann, M.; Hämäläinen, M.; Vrang, L.; Oberg, B.; Danielson, U.H. Screening for NNRTIs with slow dissociation and high affinity for a panel of HIV-1 RT variants. J. Biomol. Screen. 2009, 14, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Elleder, D.; Baiga, T.J.; Russell, R.L.; Naughton, J.A.; Hughes, S.H.; Noel, J.P.; Young, J.A. Identification of a 3-aminoimidazo[1,2-a]pyridine inhibitor of HIV-1 reverse transcriptase. Virol. J. 2012, 9, 305. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, D.; Park, C.; So, W.; Jo, M.; Ok, T.; Kwon, J.; Kong, S.; Jo, S.; Kim, Y.; et al. Discovery of Phenylaminopyridine Derivatives as Novel HIV-1 Non-nucleoside Reverse Transcriptase Inhibitors. ACS Med. Chem. Lett. 2012, 3, 678–682. [Google Scholar] [CrossRef]
- Huang, F.; Han, X.; Xiao, X.; Zhou, J. Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development. Molecules 2022, 27, 7728. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Hwang, Y.S.; Kim, M.; Park, S.B. Recent advances in the development of covalent inhibitors. RSC Med. Chem. 2021, 12, 1037–1045. [Google Scholar] [CrossRef]
- Gao, P.; Song, S.; Frutos-Beltrán, E.; Li, W.; Sun, B.; Kang, D.; Zou, J.; Zhang, J.; Pannecouque, C.; De Clercq, E.; et al. Novel indolylarylsulfone derivatives as covalent HIV-1 reverse transcriptase inhibitors specifically targeting the drug-resistant mutant Y181C. Bioorg. Med. Chem. 2021, 30, 115927. [Google Scholar] [CrossRef]
- Ippolito, J.A.; Niu, H.; Bertoletti, N.; Carter, Z.J.; Jin, S.; Spasov, K.A.; Cisneros, J.A.; Valhondo, M.; Cutrona, K.J.; Anderson, K.S.; et al. Covalent Inhibition of Wild-Type HIV-1 Reverse Transcriptase Using a Fluorosulfate Warhead. ACS Med. Chem. Lett. 2021, 12, 249–255. [Google Scholar] [CrossRef]
- Zhou, Z.; Meng, B.; An, J.; Zhao, F.; Sun, Y.; Zeng, D.; Wang, W.; Gao, S.; Xia, Y.; Dun, C.; et al. Covalently Targeted Highly Conserved Tyr318 to Improve the Drug Resistance Profiles of HIV-1 NNRTIs: A Proof-of-Concept Study. Int. J. Mol. Sci. 2023, 24, 1215. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, T.; Kang, D.; Huo, Z.; Wu, G.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; Zhan, P.; Liu, X. Discovery of novel diarylpyrimidines as potent HIV-1 NNRTIs by investigating the chemical space of a less explored “hydrophobic channel”. Org. Biomol. Chem. 2018, 16, 1014–1028. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, T.; Wu, G.; Kang, D.; Fu, Z.; Wang, Z.; De Clercq, E.; Pannecouque, C.; Zhan, P.; Liu, X. Targeting the hydrophobic channel of NNIBP: Discovery of novel 1,2,3-triazole-derived diarylpyrimidines as novel HIV-1 NNRTIs with high potency against wild-type and K103N mutant virus. Org. Biomol. Chem. 2019, 17, 3202–3217. [Google Scholar] [CrossRef]
- Kang, D.; Feng, D.; Ginex, T.; Zou, J.; Wei, F.; Zhao, T.; Huang, B.; Sun, Y.; Desta, S.; De Clercq, E.; et al. Exploring the hydrophobic channel of NNIBP leads to the discovery of novel piperidine-substituted thiophene[3,2-d]pyrimidine derivatives as potent HIV-1 NNRTIs. Acta Pharm. Sin. B 2020, 10, 878–894. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kang, D.; Feng, D.; Cherukupalli, S.; Jiang, X.; Fu, Z.; De Clercq, E.; Pannecouque, C.; Liu, X.; Zhan, P. Targeting dual tolerant regions of binding pocket: Discovery of novel morpholine-substituted diarylpyrimidines as potent HIV-1 NNRTIs with significantly improved water solubility. Eur. J. Med. Chem. 2020, 206, 112811. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Kang, D.; Da, F.; Zhang, T.; Li, P.; Zhang, B.; De Clercq, E.; Pannecouque, C.; Zhan, P.; Liu, X. Identification of novel potent HIV-1 inhibitors by exploiting the tolerant regions of the NNRTIs binding pocket. Eur. J. Med. Chem. 2021, 214, 113204. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Huang, B.; Olotu, F.A.; Li, J.; Kang, D.; Wang, Z.; De Clercq, E.; Soliman, M.E.S.; Pannecouque, C.; Liu, X.; et al. Exploiting the tolerant region I of the non-nucleoside reverse transcriptase inhibitor (NNRTI) binding pocket. Part 2: Discovery of diarylpyrimidine derivatives as potent HIV-1 NNRTIs with high Fsp(3) values and favorable drug-like properties. Eur. J. Med. Chem. 2021, 213, 113051. [Google Scholar] [CrossRef]
- Mahboubi-Rabbani, M.; Abbasi, M.; Hajimahdi, Z.; Zarghi, A. HIV-1 Reverse Transcriptase/Integrase Dual Inhibitors: A Review of Recent Advances and Structure-activity Relationship Studies. Iran. J. Pharm. Res. 2021, 20, 333–369. [Google Scholar] [CrossRef] [PubMed]
- Hazuda, D.J.; Felock, P.; Witmer, M.; Wolfe, A.; Stillmock, K.; Grobler, J.A.; Espeseth, A.; Gabryelski, L.; Schleif, W.; Blau, C.; et al. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000, 287, 646–650. [Google Scholar] [CrossRef]
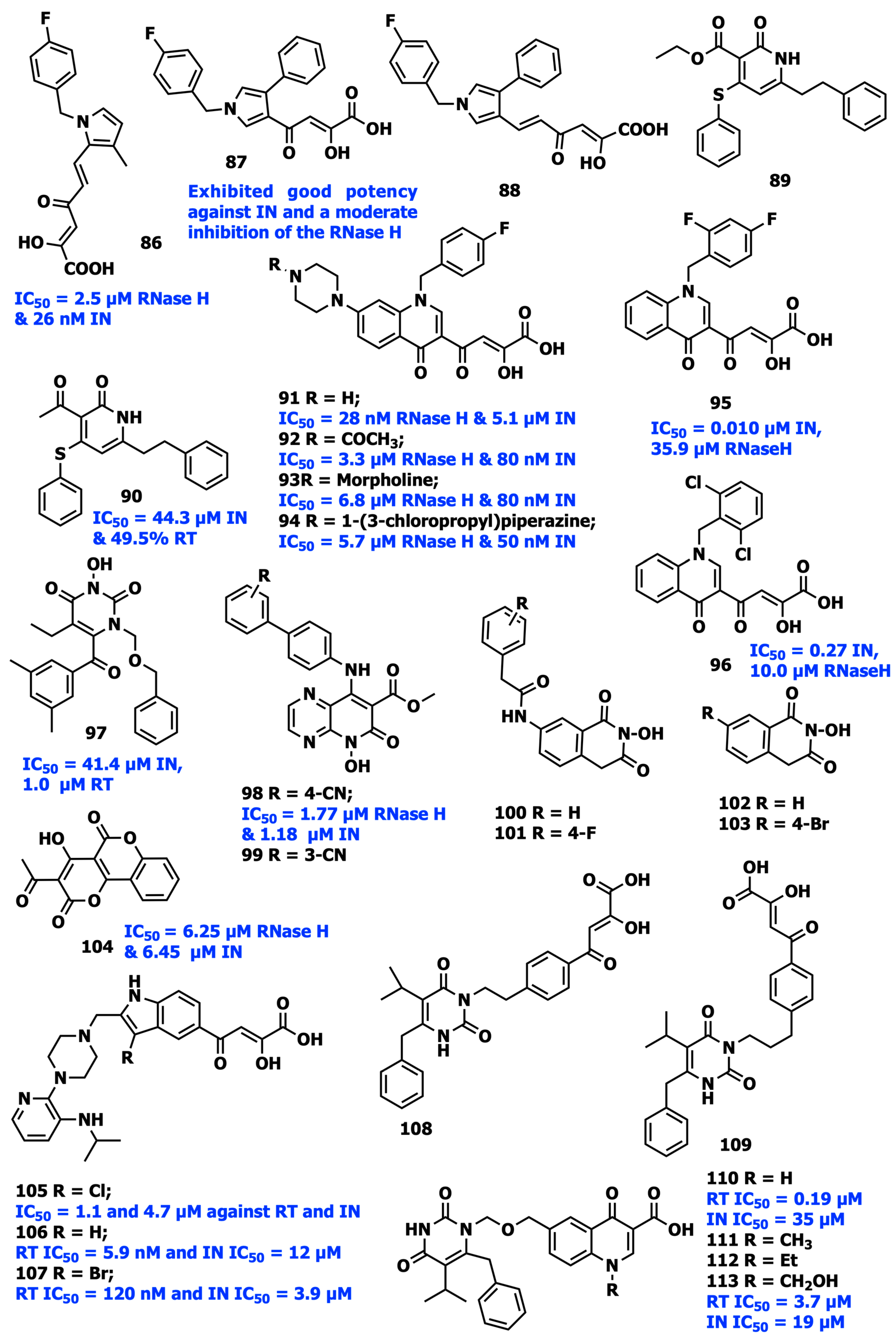
- Costi, R.; Métifiot, M.; Esposito, F.; Cuzzucoli Crucitti, G.; Pescatori, L.; Messore, A.; Scipione, L.; Tortorella, S.; Zinzula, L.; Novellino, E.; et al. 6-(1-Benzyl-1H-pyrrol-2-yl)-2,4-dioxo-5-hexenoic acids as dual inhibitors of recombinant HIV-1 integrase and ribonuclease H, synthesized by a parallel synthesis approach. J. Med. Chem. 2013, 56, 8588–8598. [Google Scholar] [CrossRef]
- Tian, C.; Liu, J.; Fan, N.; Yang, Q.; Sheng, T.; Wang, X.; Hao, Y.; Guo, Y.; Cao, Y.; Zhang, Z. Design, synthesis and activity evaluation of novel pyridinone derivatives as anti-HIV-1 dual (RT/IN) inhibitors. J. Chin. Pharm. Sci. 2017, 26, 31–44. [Google Scholar] [CrossRef]
- Costi, R.; Métifiot, M.; Chung, S.; Cuzzucoli Crucitti, G.; Maddali, K.; Pescatori, L.; Messore, A.; Madia, V.N.; Pupo, G.; Scipione, L.; et al. Basic quinolinonyl diketo acid derivatives as inhibitors of HIV integrase and their activity against RNase H function of reverse transcriptase. J. Med. Chem. 2014, 57, 3223–3234. [Google Scholar] [CrossRef]
- Pescatori, L.; Métifiot, M.; Chung, S.; Masoaka, T.; Cuzzucoli Crucitti, G.; Messore, A.; Pupo, G.; Madia, V.N.; Saccoliti, F.; Scipione, L.; et al. N-Substituted Quinolinonyl Diketo Acid Derivatives as HIV Integrase Strand Transfer Inhibitors and Their Activity against RNase H Function of Reverse Transcriptase. J. Med. Chem. 2015, 58, 4610–4623. [Google Scholar] [CrossRef]
- Das, K.; Lewi, P.J.; Hughes, S.H.; Arnold, E. Crystallography and the design of anti-AIDS drugs: Conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog. Biophys. Mol. Biol. 2005, 88, 209–231. [Google Scholar] [CrossRef]
- Chen, W.; Zhan, P.; Wu, J.; Li, Z.; Liu, X. The development of HEPT-type HIV non-nucleoside reverse transcriptase inhibitors and its implications for DABO family. Curr. Pharm. Des. 2012, 18, 4165–4186. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Maddali, K.; Dreis, C.D.; Sham, Y.Y.; Vince, R.; Pommier, Y.; Wang, Z. 6-Benzoyl-3-hydroxypyrimidine-2,4-diones as dual inhibitors of HIV reverse transcriptase and integrase. Bioorg. Med. Chem. Lett. 2011, 21, 2400–2402. [Google Scholar] [CrossRef]
- Sun, L.; Gao, P.; Dong, G.; Zhang, X.; Cheng, X.; Ding, X.; Wang, X.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; et al. 5-Hydroxypyrido[2,3-b]pyrazin-6(5H)-one derivatives as novel dual inhibitors of HIV-1 reverse transcriptase-associated ribonuclease H and integrase. Eur. J. Med. Chem. 2018, 155, 714–724. [Google Scholar] [CrossRef]
- Billamboz, M.; Bailly, F.; Barreca, M.L.; De Luca, L.; Mouscadet, J.F.; Calmels, C.; Andréola, M.L.; Witvrouw, M.; Christ, F.; Debyser, Z.; et al. Design, synthesis, and biological evaluation of a series of 2-hydroxyisoquinoline-1,3(2H,4H)-diones as dual inhibitors of human immunodeficiency virus type 1 integrase and the reverse transcriptase RNase H domain. J. Med. Chem. 2008, 51, 7717–7730. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Ambrosio, F.A.; Maleddu, R.; Costa, G.; Rocca, R.; Maccioni, E.; Catalano, R.; Romeo, I.; Eleftheriou, P.; Karia, D.C.; et al. Chromenone derivatives as a versatile scaffold with dual mode of inhibition of HIV-1 reverse transcriptase-associated Ribonuclease H function and integrase activity. Eur. J. Med. Chem. 2019, 182, 111617. [Google Scholar] [CrossRef]
- Gu, S.X.; Xue, P.; Ju, X.L.; Zhu, Y.Y. Advances in rationally designed dual inhibitors of HIV-1 reverse transcriptase and integrase. Bioorg. Med. Chem. 2016, 24, 5007–5016. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Vince, R. Design and synthesis of dual inhibitors of HIV reverse transcriptase and integrase: Introducing a diketoacid functionality into delavirdine. Bioorg. Med. Chem. 2008, 16, 3587–3595. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, J.; Salomon, C.E.; Dreis, C.D.; Vince, R. Pharmacophore and structure-activity relationships of integrase inhibition within a dual inhibitor scaffold of HIV reverse transcriptase and integrase. Bioorg. Med. Chem. 2010, 18, 4202–4211. [Google Scholar] [CrossRef]
- Wang, Z.; Vince, R. Synthesis of pyrimidine and quinolone conjugates as a scaffold for dual inhibitors of HIV reverse transcriptase and integrase. Bioorg. Med. Chem. Lett. 2008, 18, 1293–1296. [Google Scholar] [CrossRef]
- Xavier Ruiz, F.; Arnold, E. Evolving understanding of HIV-1 reverse transcriptase structure, function, inhibition, and resistance. Curr. Opin. Struct. Biol. 2020, 61, 113–123. [Google Scholar] [CrossRef]
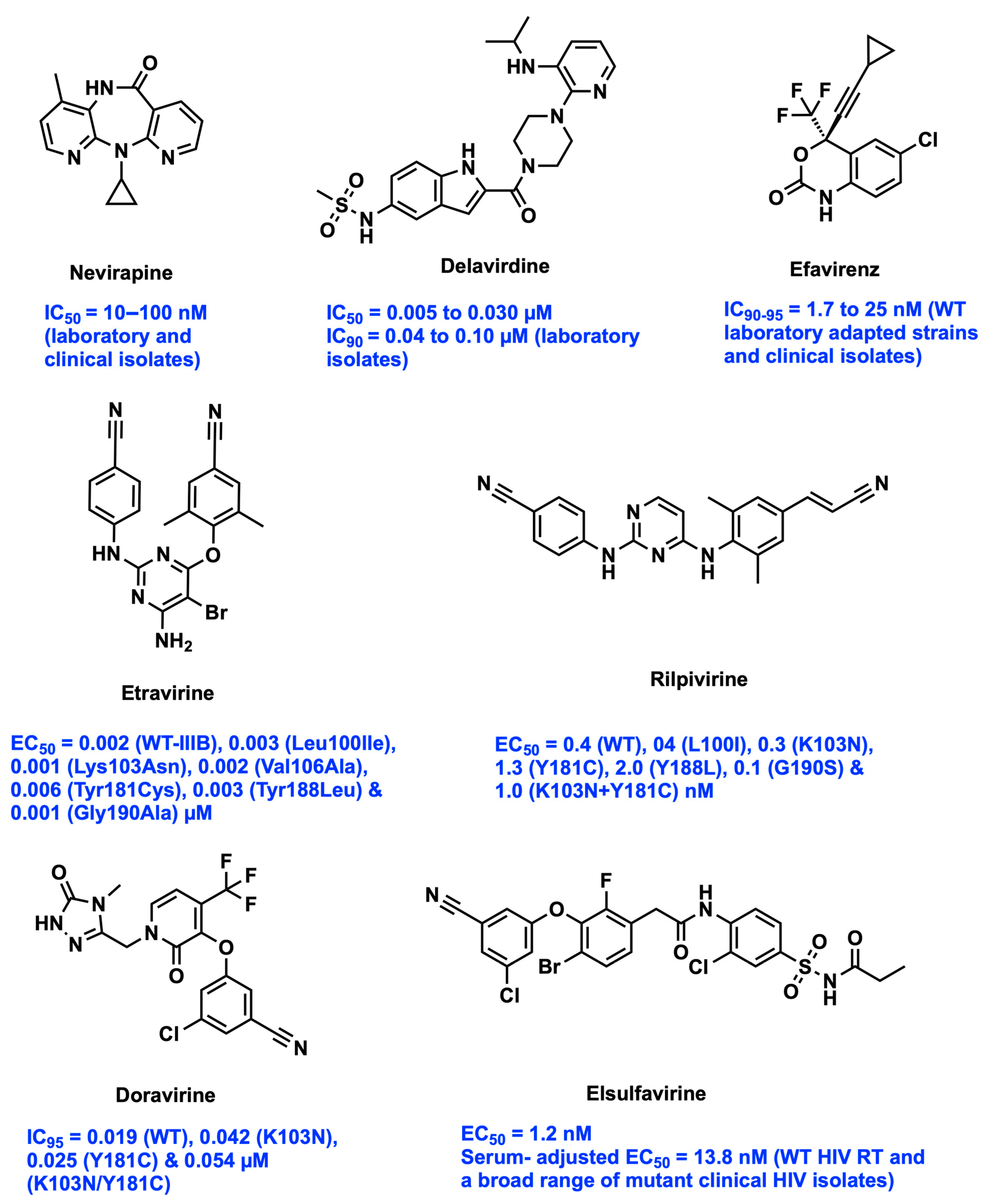
- van Heeswijk, R.P.; Veldkamp, A.I.; Mulder, J.W.; Meenhorst, P.L.; Wit, F.W.; Lange, J.M.; Danner, S.A.; Foudraine, N.A.; Kwakkelstein, M.O.; Reiss, P.; et al. The steady-state pharmacokinetics of nevirapine during once daily and twice daily dosing in HIV-1-infected individuals. AIDS 2000, 14, F77–F82. [Google Scholar] [CrossRef] [PubMed]
- Jayaweera, D.; Dilanchian, P. New therapeutic landscape of NNRTIs for treatment of HIV: A look at recent data. Expert Opin. Pharmacother. 2012, 13, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Parienti, J.J.; Peytavin, G. Nevirapine once daily: Pharmacology, metabolic profile and efficacy data of the new extended-release formulation. Expert Opin. Drug Metab. Toxicol. 2011, 7, 495–503. [Google Scholar] [CrossRef]
- Scott, L.J.; Perry, C.M. Delavirdine: A review of its use in HIV infection. Drugs 2000, 60, 1411–1444. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.F.; Neiner, A.; Kharasch, E.D. Efavirenz Metabolism: Influence of Polymorphic CYP2B6 Variants and Stereochemistry. Drug Metab. Dispos. Biol. Fate Chem. 2019, 47, 1195–1205. [Google Scholar] [CrossRef]
- Fulco, P.P.; McNicholl, I.R. Etravirine and rilpivirine: Nonnucleoside reverse transcriptase inhibitors with activity against human immunodeficiency virus type 1 strains resistant to previous nonnucleoside agents. Pharmacotherapy 2009, 29, 281–294. [Google Scholar] [CrossRef]
- Danjuma, M.I. Pharmacokinetics of HIV non-nucleoside reverse-transcriptase inhibitors. Future Med. 2009, 3, 625–632. [Google Scholar] [CrossRef]
- Usach, I.; Melis, V.; Peris, J.-E. Non-nucleoside reverse transcriptase inhibitors: A review on pharmacokinetics, pharmacodynamics, safety and tolerability. J. Int. AIDS Soc. 2013, 16, 1–14. [Google Scholar] [CrossRef]
- Zdanowicz, M.M. The pharmacology of HIV drug resistance. Am. J. Pharm. Educ. 2006, 70, 100. [Google Scholar] [CrossRef]
- Rai, M.A.; Pannek, S.; Fichtenbaum, C.J. Emerging reverse transcriptase inhibitors for HIV-1 infection. Expert Opin. Emerg. Drugs 2018, 23, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.; Moss, C.E.; Marzolini, C.; Khoo, S. Clinical Pharmacodynamics, Pharmacokinetics, and Drug Interaction Profile of Doravirine. Clin. Pharmacokinet. 2019, 58, 1553–1565. [Google Scholar] [CrossRef] [PubMed]
- Talwani, R.; Temesgen, Z. Doravirine: A new non-nucleoside reverse transcriptase inhibitor for the treatment of HIV infection. Drugs Today 2020, 56, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.T.; Xiao, M.A.; Principe, M.A.; Wong, A.; Mesplède, T. Pharmaceutical, clinical, and resistance information on doravirine, a novel non-nucleoside reverse transcriptase inhibitor for the treatment of HIV-1 infection. Drugs Context 2020, 9, 2019-11-4. [Google Scholar] [CrossRef]
- Kravchenko, A.; Orlova-Morozova, E.; Shimonova, T.; Kozyrev, O.; Nagimova, F.; Zakharova, N.G.; Ivanova, E.; Kuimova, U.; Efremova, O.; Sonin, D.; et al. Efficacy and safety of novel russian non-nucleoside reverse transcriptase inhibitor elsulfavirine in combination with tenofovir/emtricitabine multicenter comparative trial vs efavirenz in naive HIV patients. J. Infectology 2018, 10, 76–82. [Google Scholar] [CrossRef]
- Marrazzo, J.M.; del Rio, C.; Holtgrave, D.R.; Cohen, M.S.; Kalichman, S.C.; Mayer, K.H.; Montaner, J.S.G.; Wheeler, D.P.; Grant, R.M.; Grinsztejn, B.; et al. HIV prevention in clinical care settings: 2014 recommendations of the International Antiviral Society-USA Panel. JAMA 2014, 312, 390–409. [Google Scholar] [CrossRef] [PubMed]
- Saag, M.S.; Benson, C.A.; Gandhi, R.T.; Hoy, J.F.; Landovitz, R.J.; Mugavero, M.J.; Sax, P.E.; Smith, D.M.; Thompson, M.A.; Buchbinder, S.P.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2018 Recommendations of the International Antiviral Society-USA Panel. JAMA 2018, 320, 379–396. [Google Scholar] [CrossRef]
- Caplan, M.R.; Daar, E.S.; Corado, K.C. Next generation fixed dose combination pharmacotherapies for treating HIV. Expert Opin. Pharmacother. 2018, 19, 589–596. [Google Scholar] [CrossRef]
- Aboud, M.; Orkin, C.; Podzamczer, D.; Bogner, J.R.; Baker, D.; Khuong-Josses, M.A.; Parks, D.; Angelis, K.; Kahl, L.P.; Blair, E.A.; et al. Efficacy and safety of dolutegravir-rilpivirine for maintenance of virological suppression in adults with HIV-1: 100-week data from the randomised, open-label, phase 3 SWORD-1 and SWORD-2 studies. Lancet HIV 2019, 6, e576–e587. [Google Scholar] [CrossRef]
- Bernardini, C.; Maggiolo, F. Triple-combination rilpivirine, emtricitabine, and tenofovir (Complera™/Eviplera™) in the treatment of HIV infection. Patient Prefer. Adherence 2013, 7, 531–542. [Google Scholar] [CrossRef]
- Deeks, E.D.; Perry, C.M. Efavirenz/emtricitabine/tenofovir disoproxil fumarate single-tablet regimen (Atripla®): A review of its use in the management of HIV infection. Drugs 2010, 70, 2315–2338. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Perno, C.F.; Mallon, P.W.; Behrens, G.; Corbeau, P.; Routy, J.P.; Darcis, G. Two-drug vs. three-drug combinations for HIV-1: Do we have enough data to make the switch? HIV Med. 2019, 20 (Suppl. 4), 2–12. [Google Scholar] [CrossRef]
- Dickinson, L.; Amin, J.; Else, L.; Boffito, M.; Egan, D.; Owen, A.; Khoo, S.; Back, D.; Orrell, C.; Clarke, A.; et al. Comprehensive Pharmacokinetic, Pharmacodynamic and Pharmacogenetic Evaluation of Once-Daily Efavirenz 400 and 600 mg in Treatment-Naïve HIV-Infected Patients at 96 Weeks: Results of the ENCORE1 Study. Clin. Pharmacokinet. 2016, 55, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Das, D.; Kobayakawa, T.; Tamamura, H.; Takeuchi, H. Discovery and Development of Anti-HIV Therapeutic Agents: Progress Towards Improved HIV Medication. Curr. Top. Med. Chem. 2019, 19, 1621–1649. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Pauly, G.T.; Akram, A.; Melody, K.; Ambrose, Z.; Schneider, J.P.; Hughes, S.H. Rilpivirine and Doravirine Have Complementary Efficacies Against NNRTI-Resistant HIV-1 Mutants. J. Acquir. Immune Defic. Syndr. (1999) 2016, 72, 485–491. [Google Scholar] [CrossRef]
- Rock, A.E.; Lerner, J.; Badowski, M.E. Doravirine and Its Potential in the Treatment of HIV: An Evidence-Based Review of the Emerging Data. HIV AIDS 2020, 12, 201–210. [Google Scholar] [CrossRef]
- Llibre, J.M.; Hung, C.C.; Brinson, C.; Castelli, F.; Girard, P.M.; Kahl, L.P.; Blair, E.A.; Angelis, K.; Wynne, B.; Vandermeulen, K.; et al. Efficacy, safety, and tolerability of dolutegravir-rilpivirine for the maintenance of virological suppression in adults with HIV-1: Phase 3, randomised, non-inferiority SWORD-1 and SWORD-2 studies. Lancet 2018, 391, 839–849. [Google Scholar] [CrossRef]
- D’Amico, R.; Margolis, D.A. Long-acting injectable therapy: An emerging paradigm for the treatment of HIV infection. Curr. Opin. HIV AIDS 2020, 15, 13–18. [Google Scholar] [CrossRef]
- Spreen, W.R.; Margolis, D.A.; Pottage, J.C., Jr. Long-acting injectable antiretrovirals for HIV treatment and prevention. Curr. Opin. HIV AIDS 2013, 8, 565–571. [Google Scholar] [CrossRef]
- Williams, P.E.; Crauwels, H.M.; Basstanie, E.D. Formulation and pharmacology of long-acting rilpivirine. Curr. Opin. HIV AIDS 2015, 10, 233–238. [Google Scholar] [CrossRef]
- Roy, U.; Rodríguez, J.; Barber, P.; das Neves, J.; Sarmento, B.; Nair, M. The potential of HIV-1 nanotherapeutics: From in vitro studies to clinical trials. Nanomedicine 2015, 10, 3597–3609. [Google Scholar] [CrossRef] [PubMed]
- Al-Salama, Z.T. Elsulfavirine: First Global Approval. Drugs 2017, 77, 1811–1816. [Google Scholar] [CrossRef]
- Surve, D.H.; Jirwankar, Y.B.; Dighe, V.D.; Jindal, A.B. Long-Acting Efavirenz and HIV-1 Fusion Inhibitor Peptide Co-loaded Polymer-Lipid Hybrid Nanoparticles: Statistical Optimization, Cellular Uptake, and In Vivo Biodistribution. Mol. Pharm. 2020, 17, 3990–4003. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.; Antela, A.; Mills, A.; Huang, J.; Jäger, H.; Bernal, E.; Lombaard, J.; Katner, H.; Walmsley, S.; Khuong-Josses, M.A.; et al. Patient-Reported Outcomes in ATLAS and FLAIR Participants on Long-Acting Regimens of Cabotegravir and Rilpivirine over 48 Weeks. AIDS Behav. 2020, 24, 3533–3544. [Google Scholar] [CrossRef]
- Markham, A. Cabotegravir Plus Rilpivirine: First Approval. Drugs 2020, 80, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Taki, E.; Soleimani, F.; Asadi, A.; Ghahramanpour, H.; Namvar, A.; Heidary, M. Cabotegravir/Rilpivirine: The last FDA-approved drug to treat HIV. Expert Rev. Anti. Infect. Ther. 2022, 20, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Guida, M.; Farris, M.; Aquino, C.I.; Rosato, E.; Cipullo, L.M.A.; Bastianelli, C. Nexplanon Subdermal Implant: Assessment of Sexual Profile, Metabolism, and Bleeding in a Cohort of Italian Women. BioMed Res. Int. 2019, 2019, 3726957. [Google Scholar] [CrossRef]
- Barrett, S.E.; Teller, R.S.; Forster, S.P.; Li, L.; Mackey, M.A.; Skomski, D.; Yang, Z.; Fillgrove, K.L.; Doto, G.J.; Wood, S.L.; et al. Extended-Duration MK-8591-Eluting Implant as a Candidate for HIV Treatment and Prevention. Antimicrob. Agents Chemother. 2018, 62, e01058-18. [Google Scholar] [CrossRef]
- Schürmann, D.; Rudd, D.J.; Zhang, S.; De Lepeleire, I.; Robberechts, M.; Friedman, E.; Keicher, C.; Hüser, A.; Hofmann, J.; Grobler, J.A.; et al. Safety, pharmacokinetics, and antiretroviral activity of islatravir (ISL, MK-8591), a novel nucleoside reverse transcriptase translocation inhibitor, following single-dose administration to treatment-naive adults infected with HIV-1: An open-label, phase 1b, consecutive-panel trial. Lancet HIV 2020, 7, e164–e172. [Google Scholar] [CrossRef]
- Beloor, J.; Kudalkar, S.N.; Buzzelli, G.; Yang, F.; Mandl, H.K.; Rajashekar, J.K.; Spasov, K.A.; Jorgensen, W.L.; Saltzman, W.M.; Anderson, K.S.; et al. Long-acting and extended-release implant and nanoformulations with a synergistic antiretroviral two-drug combination controls HIV-1 infection in a humanized mouse model. Bioeng. Transl. Med. 2022, 7, e10237. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Brand | Drug | Clinical Status | Administration |
|---|---|---|---|
| Cabenuva | Cabotegravir + Rilpivirine | Approved | Suspension Injection |
| Elpida | Elsulfavirine | Preclinical | Oral formulation [143] |
| - | Efavirenz (Polymer lipids with nanoparticles co-loaded with Enfuvirtide) | Preclinical | Subcutaneous [144] |
| Brand | Edurant | Rekambys | Vocabria | - |
|---|---|---|---|---|
| Drug | Rilpivirine | Rilpivirine | Cabotegravir | Cabotegravir |
| Route of Administration | Oral | Injection | Oral | Injection |
| Strength | 25mg | 900mg i; 600mg m | 30mg | 600mg i; 400mg m |
| Tmax | 4–5 h | 3 to 4 days | 3 h | 7 days |
| Blood-to-plasma ratio | 0.7 | 0.7 | 0.5 | 0.5 |
| t1/2, mean | 45–50 h | 13 to 28 weeks | 41 h | 5.6 to 11.5 weeks |
| Subsequent-dose pharmacokinetic | ||||
| Cmax | 247 ng/mL | 116 ng/mL | 8.1 µg/mL | 4.2 µg /mL |
| AUCtau | 3300 ngh/mL | 65603 (ngh/mL) | 146 µg/mL | 2461 µg /mL |
| Time taken | Day 7 | 44 weeks | Day 7 | 44 weeks |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanangamudi, M.; Palaniappan, S.; Kathiravan, M.K.; Namasivayam, V. Strategies in the Design and Development of Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs). Viruses 2023, 15, 1992. https://doi.org/10.3390/v15101992
Vanangamudi M, Palaniappan S, Kathiravan MK, Namasivayam V. Strategies in the Design and Development of Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs). Viruses. 2023; 15(10):1992. https://doi.org/10.3390/v15101992
Chicago/Turabian StyleVanangamudi, Murugesan, Senthilkumar Palaniappan, Muthu Kumaradoss Kathiravan, and Vigneshwaran Namasivayam. 2023. "Strategies in the Design and Development of Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs)" Viruses 15, no. 10: 1992. https://doi.org/10.3390/v15101992