
Genetic Evolution of Avian Influenza A (H9N2) Viruses Isolated from Domestic Poultry in Uganda Reveals Evidence of Mammalian Host Adaptation, Increased Virulence and Reduced Sensitivity to Baloxavir

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Whole Genome Sequencing and Sequence Identification
2.3. Sequence Analysis
2.4. Assessment of H9N2 Susceptibility to Baloxivir (BXA)
2.5. Hemagglutination Inhibition (HI) Assay
3. Results
3.1. H9N2 Detection
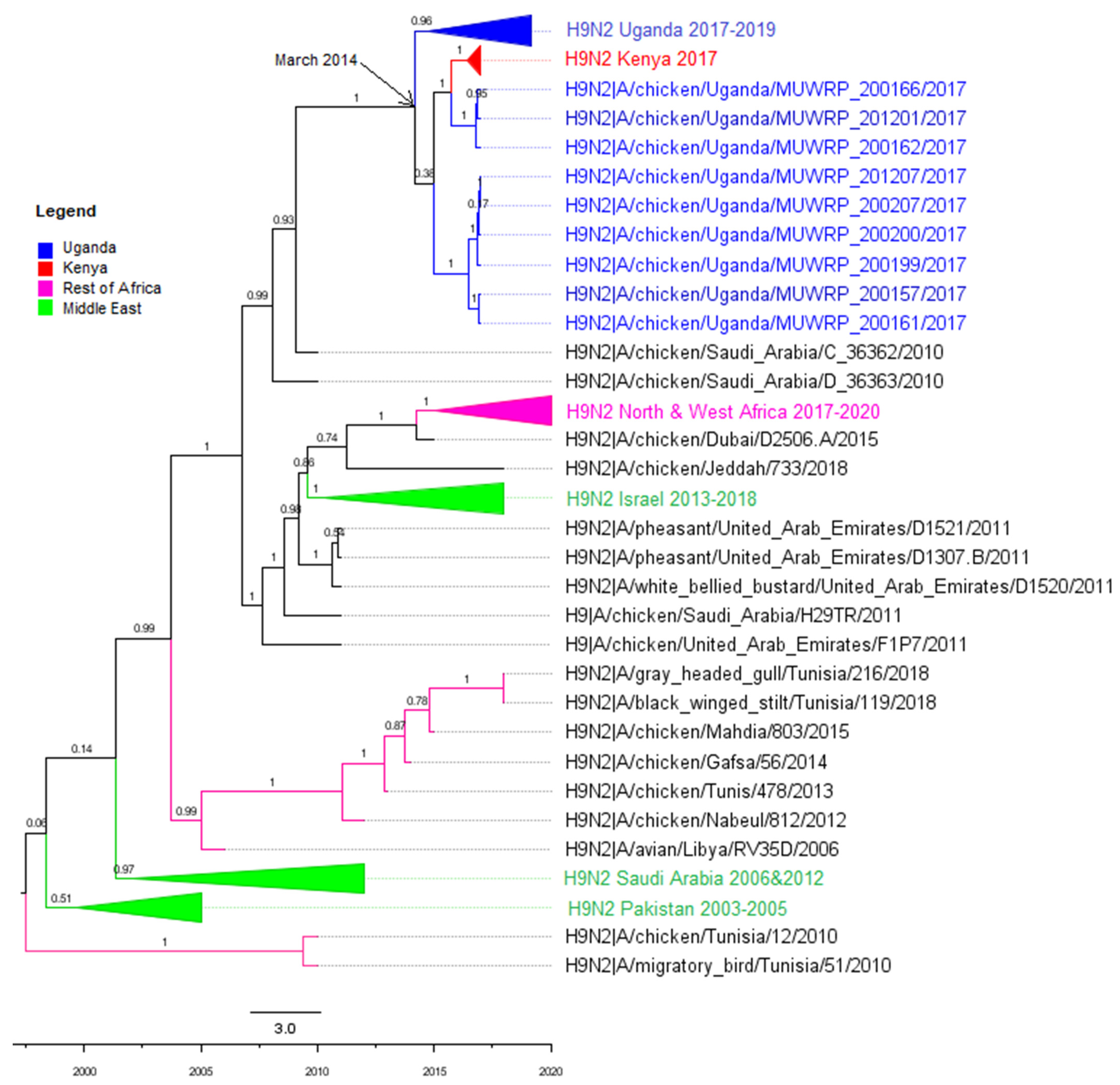
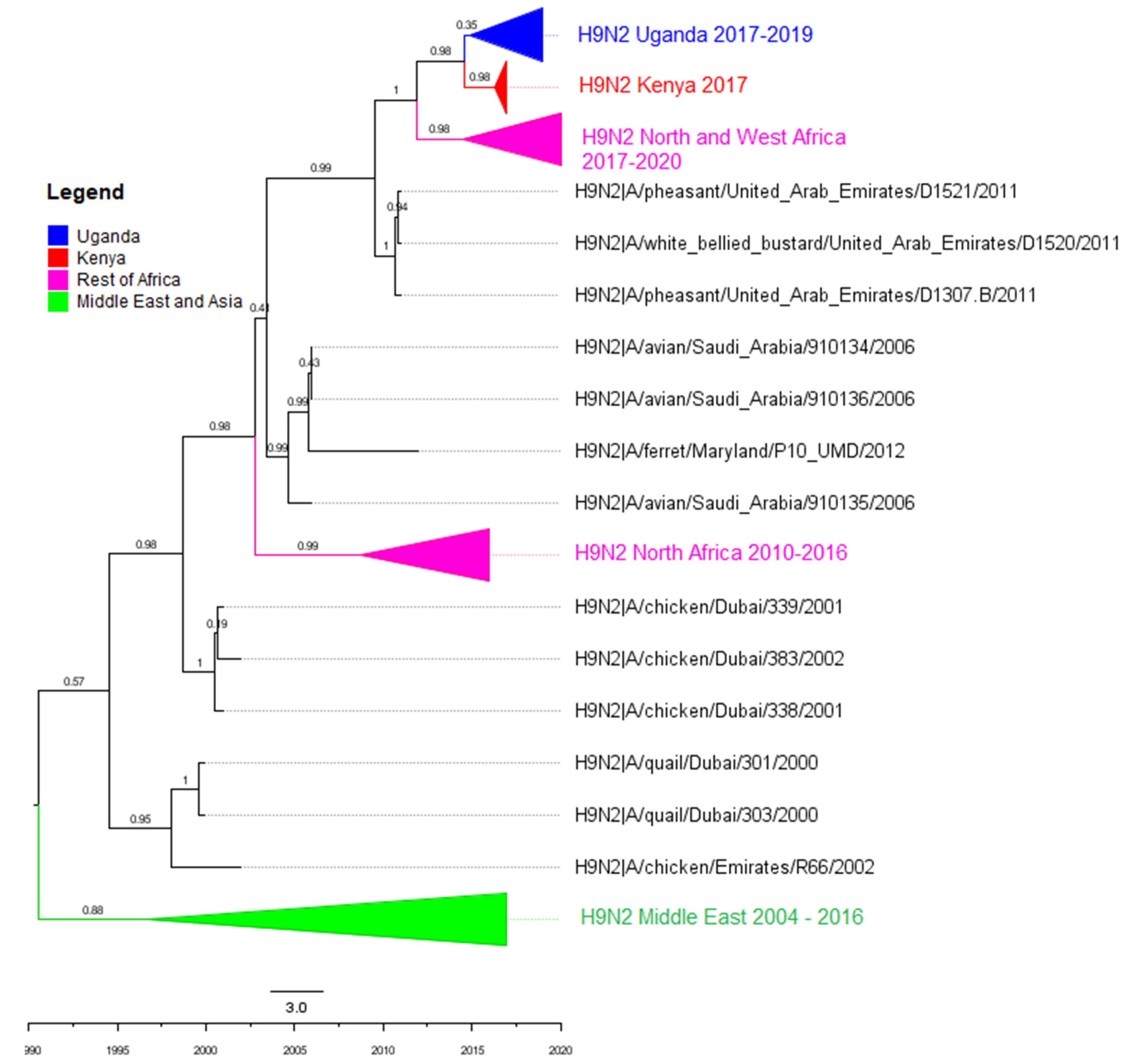
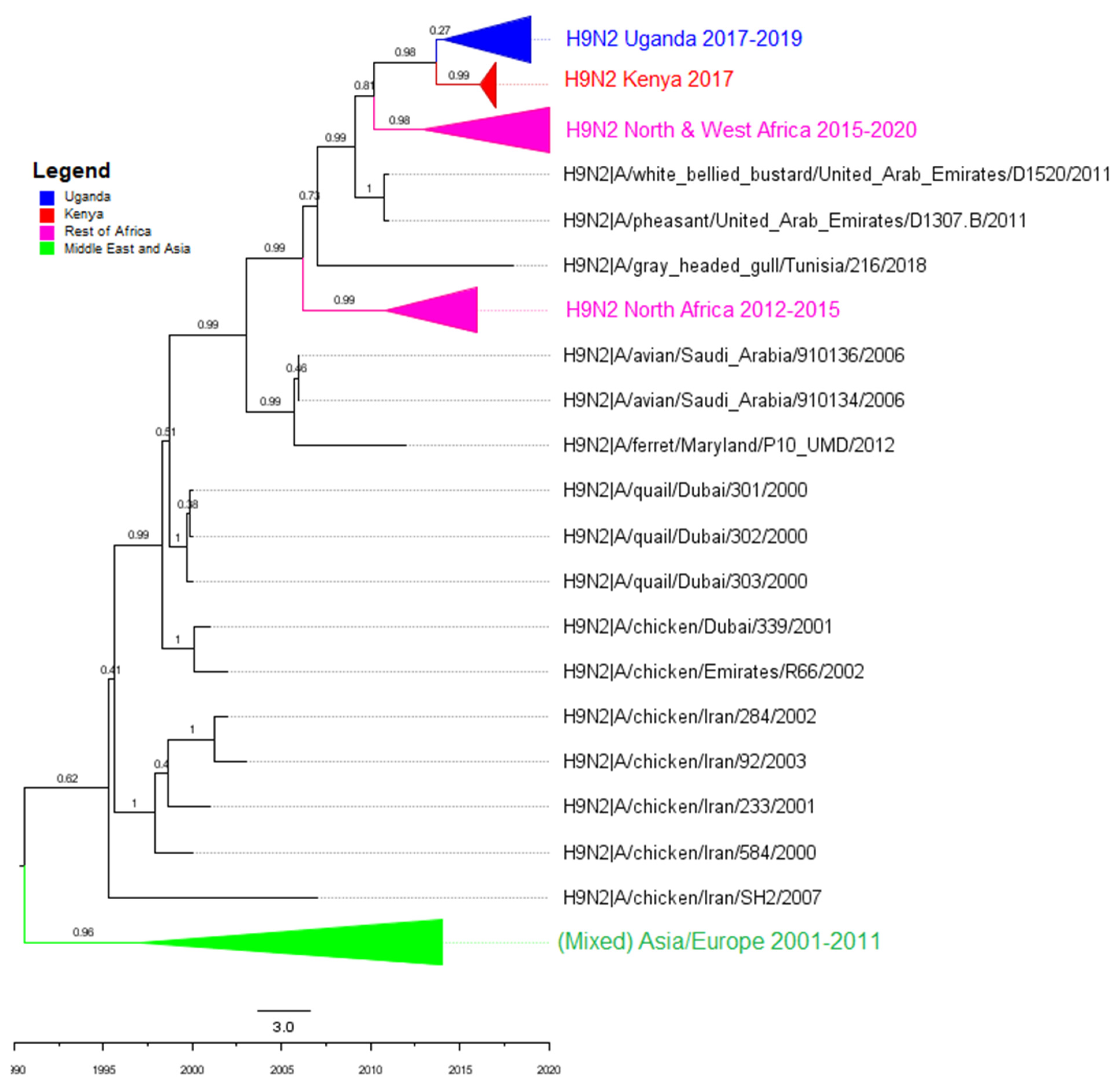
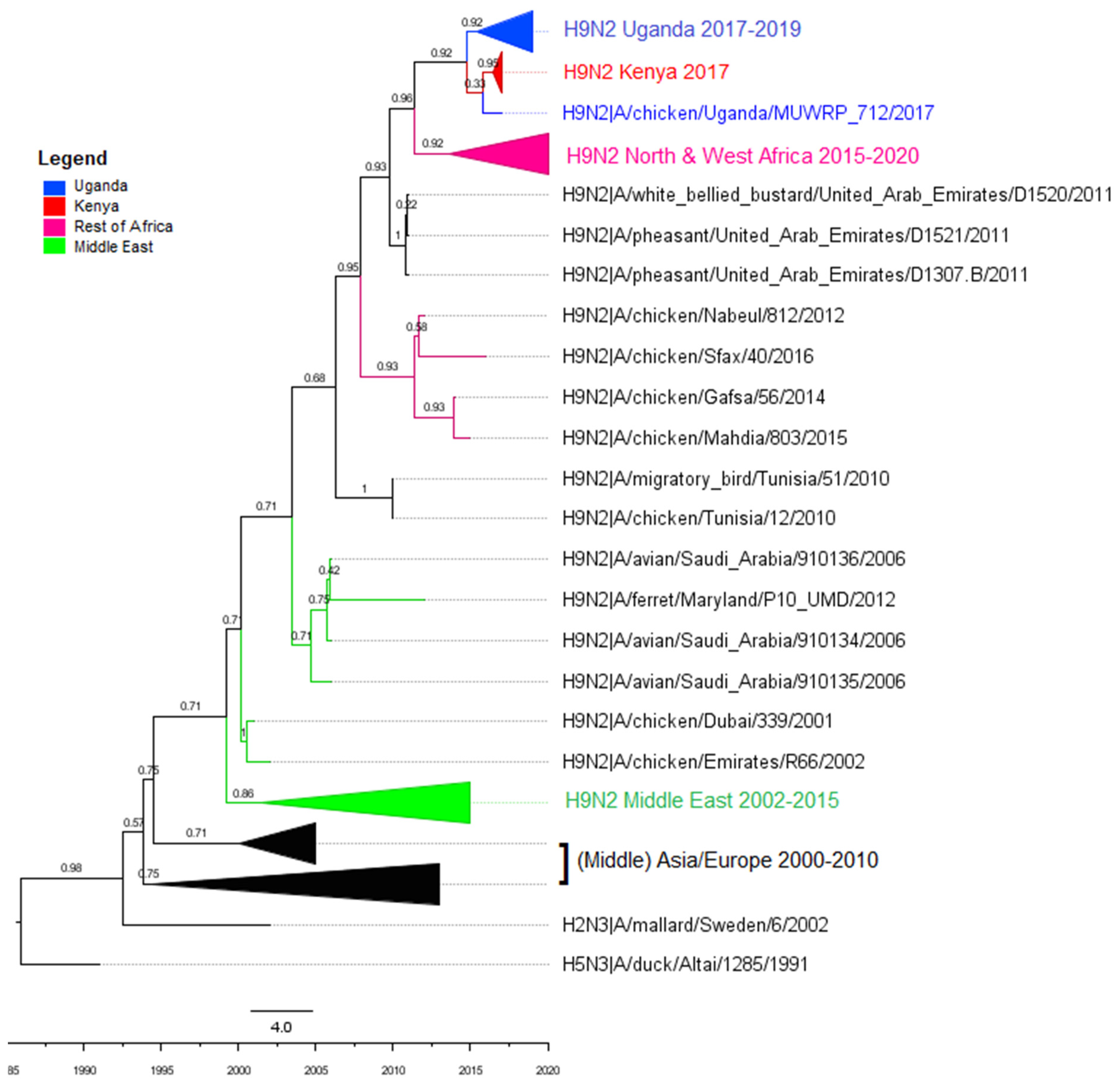
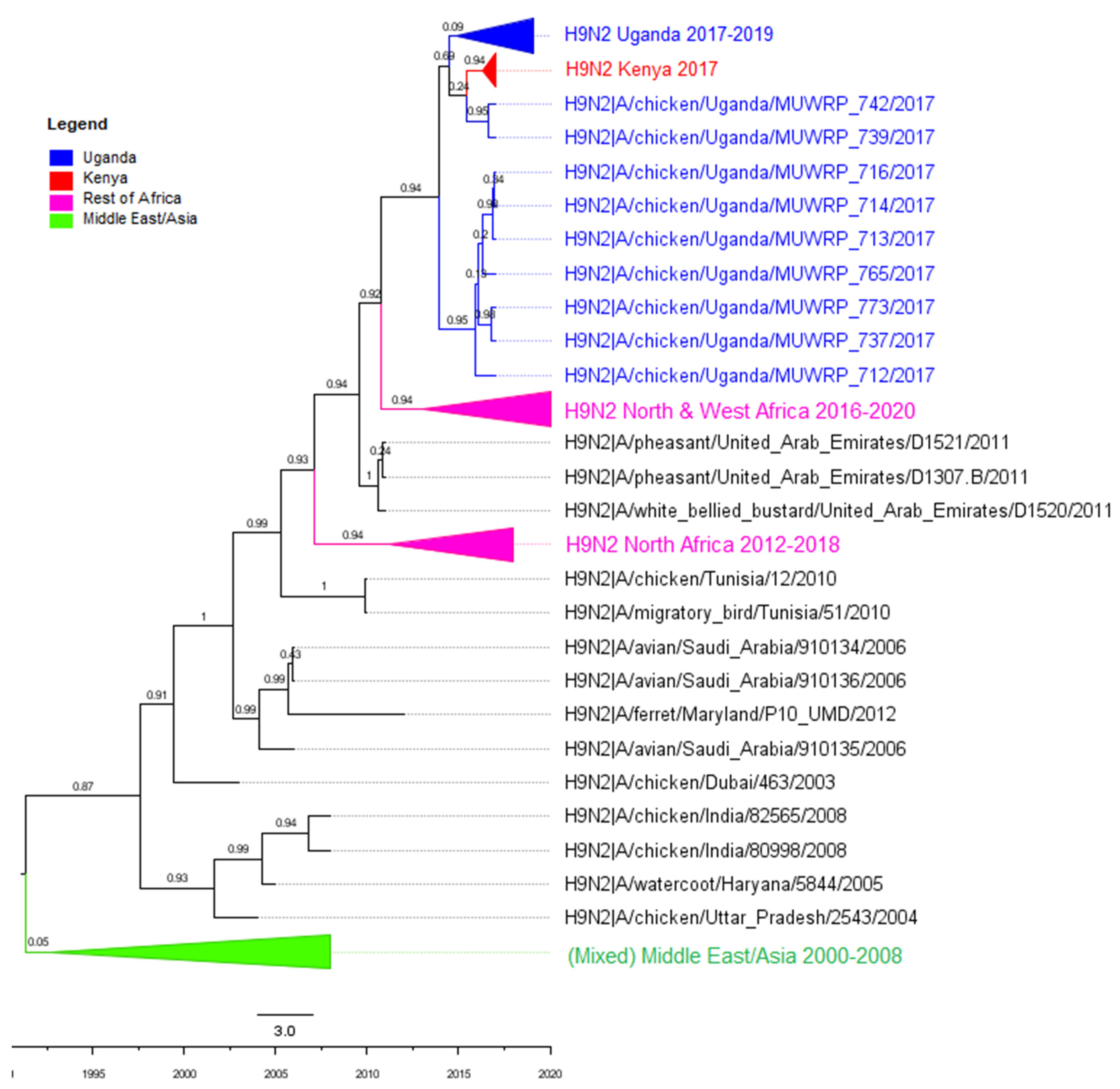
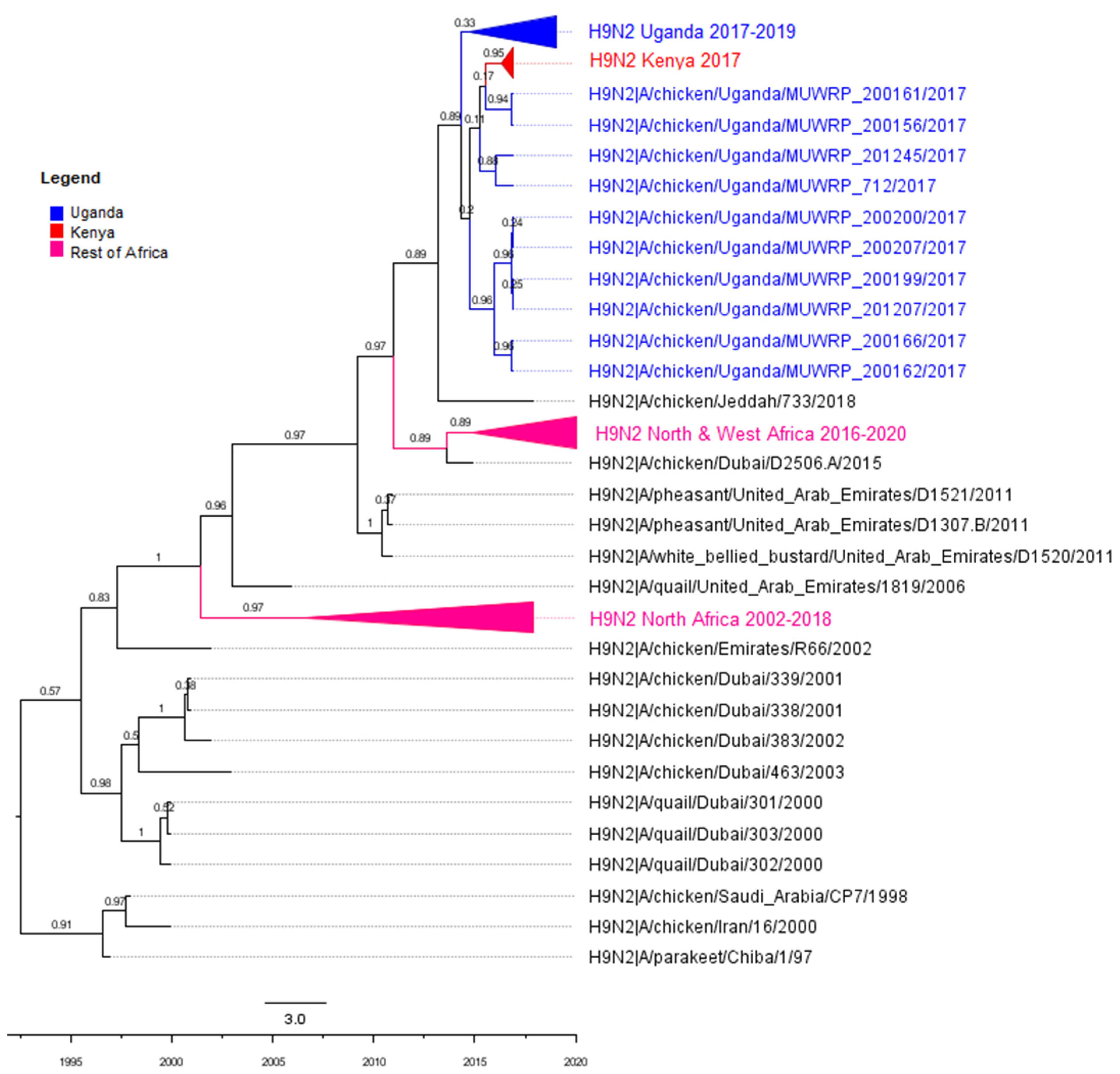
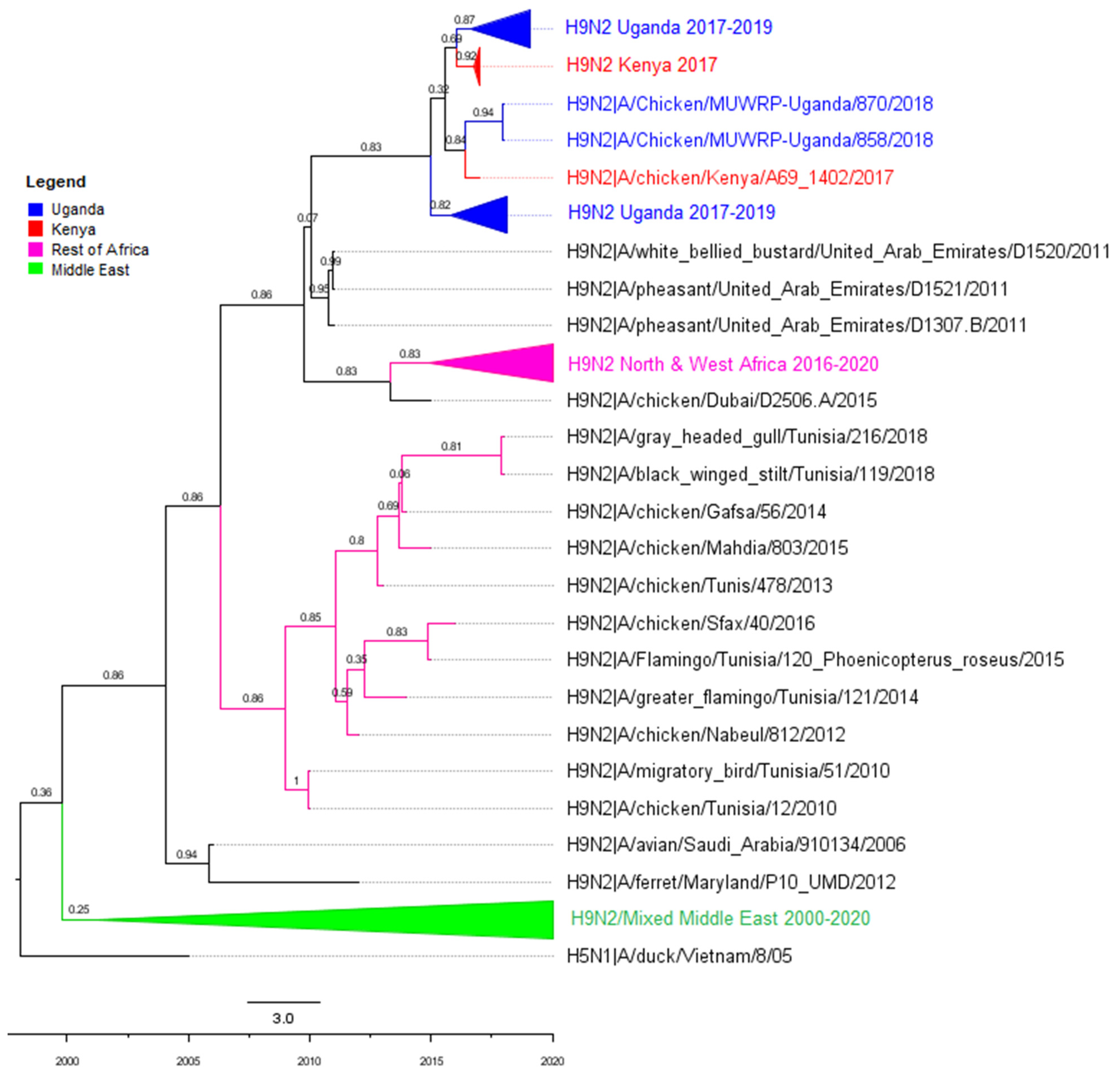
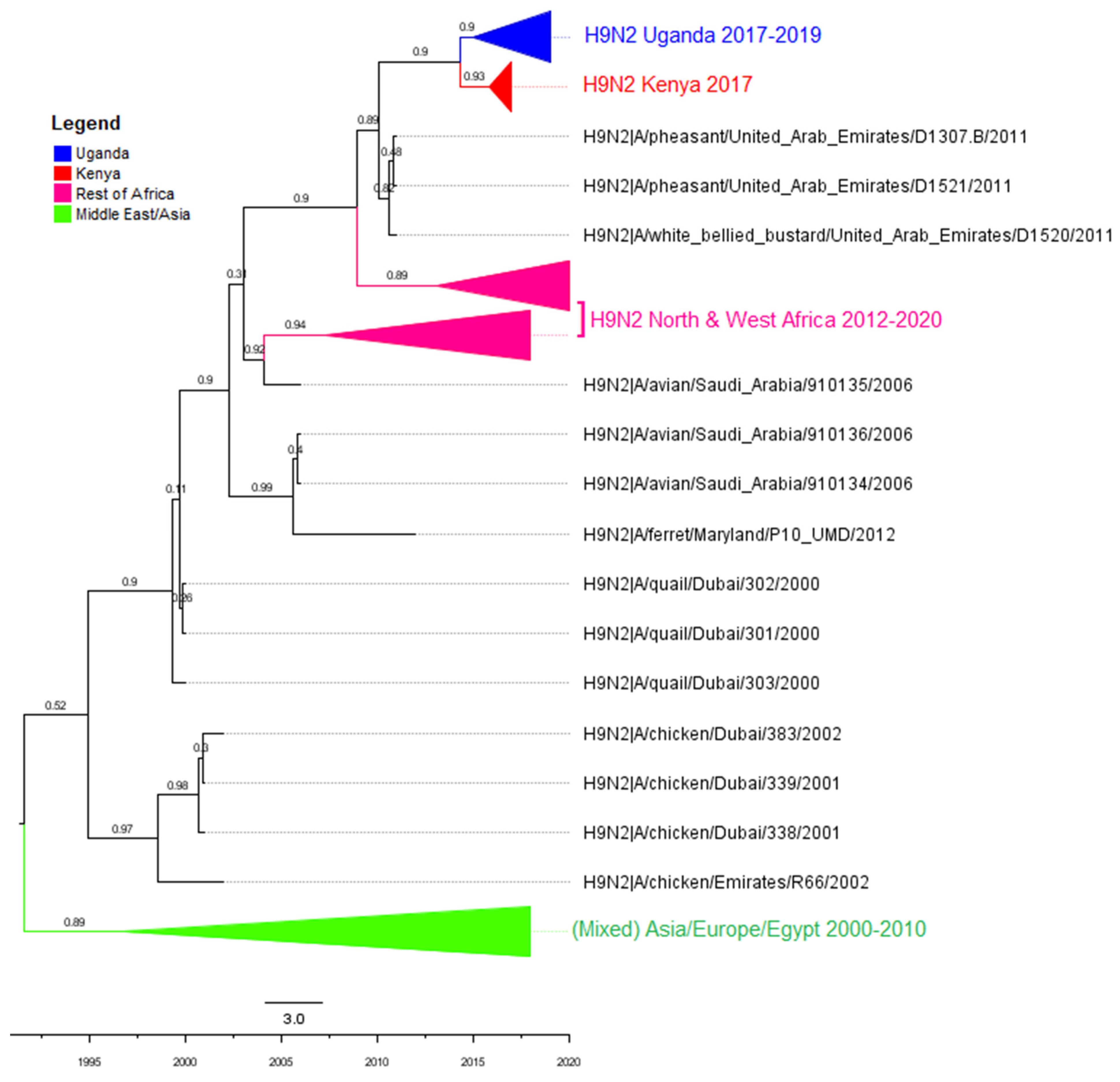
3.2. Phylogenetic Analyses
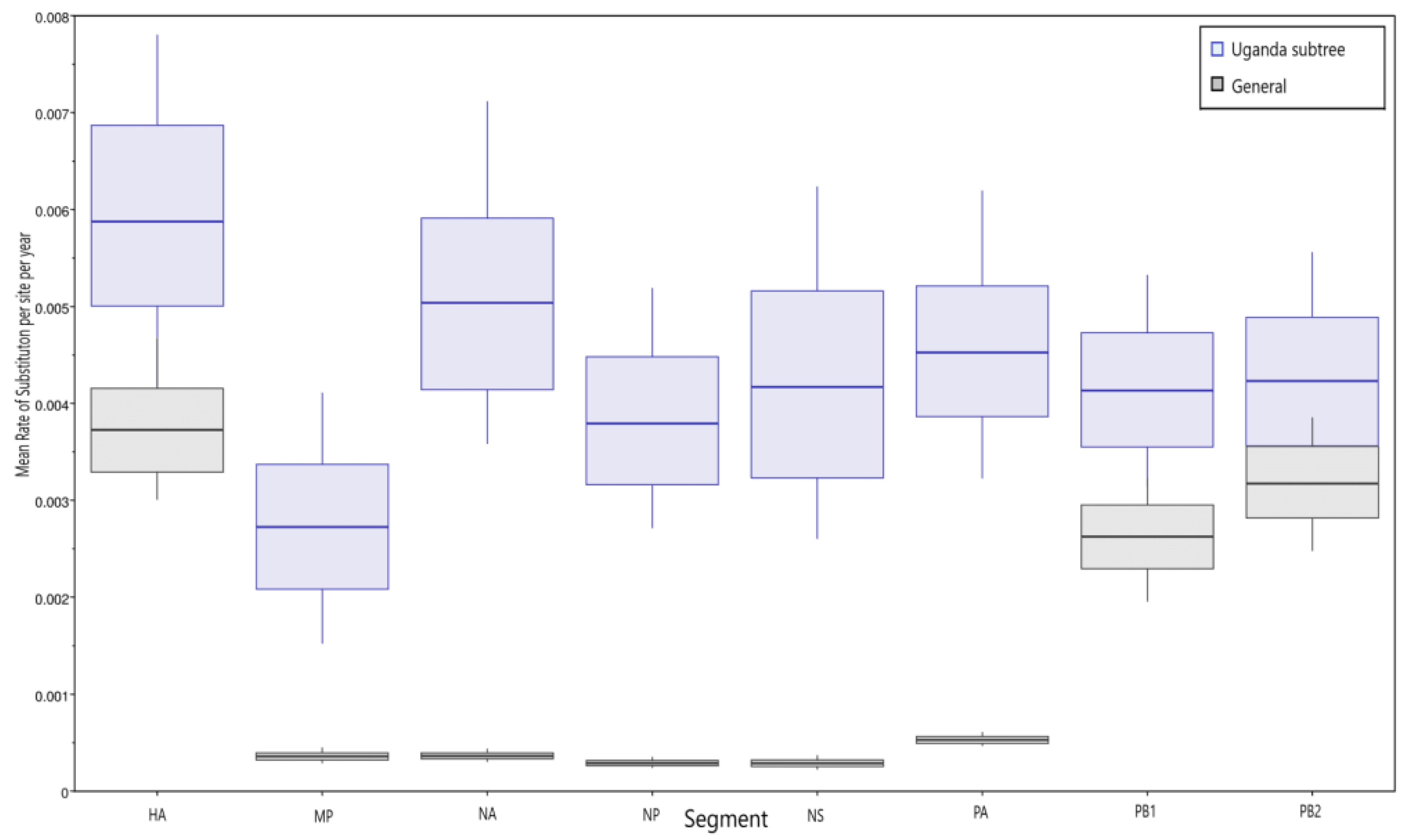
3.3. Molecular Clock Analysis
3.4. Reassortment
3.5. Molecular Marker Analysis
3.6. Antigenic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A







References
- Peacock, T.P.; James, J.; Sealy, J.E.; Iqbal, M. A Global Perspective on H9N2 Avian Influenza Virus. Viruses 2019, 11, 620. [Google Scholar] [CrossRef] [PubMed]
- Mertens, E.; Dugan, V.G.; Stockwell, T.B.; Lindsay, L.L.; Plancarte, M.; Boyce, W.M. Evaluation of Phenotypic Markers in Full Genome Sequences of Avian Influenza Isolates from California. Comp. Immunol. Microbiol. Infect. Dis. 2013, 36, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Li, X.; Goraya, M.; Wang, S.; Chen, J.-L. Evolution of Influenza A Virus by Mutation and Re-Assortment. Int. J. Mol. Sci. 2017, 18, 1650. [Google Scholar] [CrossRef] [PubMed]
- Lowen, A.C. It’s in the Mix: Reassortment of Segmented Viral Genomes. PLoS Pathog. 2018, 14, e1007200. [Google Scholar] [CrossRef]
- Rossman, J.S.; Lamb, R.A. Influenza Virus Assembly and Budding. Virology 2011, 411, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Ibrahim, M.S.; Elgendy, E.M.; Daidoji, T.; Ono, T.; Suzuki, Y.; Nakaya, T.; Matsumoto, K.; Watanabe, Y. Genetic Compatibility of Reassortants between Avian H5N1 and H9N2 Influenza Viruses with Higher Pathogenicity in Mammals. J. Virol. 2019, 93, e01969-18. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.J.; Krauss, S.; Senne, D.A.; Mo, I.P.; Lo, K.S.; Xiong, X.P.; Norwood, M.; Shortridge, K.F.; Webster, R.G.; Guan, Y. Characterization of the Pathogenicity of Members of the Newly Established H9N2 Influenza Virus Lineages in Asia. Virology 2000, 267, 279–288. [Google Scholar] [CrossRef]
- Naguib, M.M.; Ulrich, R.; Kasbohm, E.; Eng, C.L.P.; Hoffmann, D.; Grund, C.; Beer, M.; Harder, T.C. Natural Reassortants of Potentially Zoonotic Avian Influenza Viruses H5N1 and H9N2 from Egypt Display Distinct Pathogenic Phenotypes in Experimentally Infected Chickens and Ferrets. J. Virol. 2017, 91, e01300-17. [Google Scholar] [CrossRef]
- Pu, J.; Wang, S.; Yin, Y.; Zhang, G.; Carter, R.A.; Wang, J.; Xu, G.; Sun, H.; Wang, M.; Wen, C.; et al. Evolution of the H9N2 Influenza Genotype That Facilitated the Genesis of the Novel H7N9 Virus. Proc. Natl. Acad. Sci. USA 2015, 112, 548–553. [Google Scholar] [CrossRef]
- WHO. Influenza at the Human-Animal Interface Summary and Assessment 27 June 2022; World Health Organization: Geneva, Switzerland, 2022.
- Zhang, M.; Zhao, C.; Chen, H.; Teng, Q.; Jiang, L.; Feng, D.; Li, X.; Yuan, S.; Xu, J.; Zhang, X.; et al. Internal Gene Cassette From a Human-Origin H7N9 Influenza Virus Promotes the Pathogenicity of H9N2 Avian Influenza Virus in Mice. Front. Microbiol. 2020, 11, 1441. [Google Scholar] [CrossRef]
- Homme, P.J.; Easterday, B.C. Avian Influenza Virus Infection. I. Characteristics of Influenza A/Turkey/Wisconsin/1966 Virus. Avian Dis. 1970, 14, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Kaboudi, K. Low Pathogenic Avian Influenza Virus Subtype H9N2 in Poultry in North Africa: Current Status. Vet. Sci. Res. Rev. 2019, 5, 73–79. [Google Scholar] [CrossRef]
- Nagy, A.; Mettenleiter, T.C.; Abdelwhab, E.M. A Brief Summary of the Epidemiology and Genetic Relatedness of Avian Influenza H9N2 Virus in Birds and Mammals in the Middle East and North Africa. Epidemiol. Infect. 2017, 145, 3320–3333. [Google Scholar] [CrossRef]
- Kirunda, H.; Kibuuka, H.; Byaruhanga, A.; Mworozi, E.; Luswa, L.; Millard, M.; Wabwire-Mangen, F. Poor Biosecurity in Live Bird Markets in Uganda: A Potential Risk for Highly Pathogenic Avian Influenza Disease Outbreak in Poultry and Spread to Humans. Int. J. Public Health Epidemiol. 2014, 3, 67–74. [Google Scholar]
- Fusade-Boyer, M.; Djegui, F.; Batawui, K.; Byuragaba, D.K.; Jones, J.C.; Wabwire-Mangeni, F.; Erima, B.; Atim, G.; Ukuli, Q.A.; Tugume, T.; et al. Antigenic and Molecular Characterization of Low Pathogenic Avian Influenza A(H9N2) Viruses in Sub-Saharan Africa from 2017 through 2019. Emerg. Microbes Infect. 2021, 10, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Public Health Agency of Canada. Human Emerging Respiratory Pathogens Bulletin; Public Health Agency of Canada: Ottawa, ON, Canada, 2022.
- Qi, Y.; Guo, W.; Liu, C.; Li, W.; Gu, Y.; Li, S.; Chen, X. Seroprevalence of Influenza A (H9N2) Virus Infection among Humans in China: A Meta-Analysis. Microb. Pathog. 2021, 155, 104881. [Google Scholar] [CrossRef]
- Almayahi, Z.K.; Al Kindi, H.; Davies, C.T.; Al-Rawahi, B.; Al-Jardani, A.; Al-Yaqoubi, F.; Jang, Y.; Jones, J.; Barnes, J.R.; Davis, W.; et al. First Report of Human Infection with Avian Influenza A(H9N2) Virus in Oman: The Need for a One Health Approach. Int. J. Infect. Dis. 2020, 91, 169–173. [Google Scholar] [CrossRef]
- Potdar, V.; Hinge, D.; Satav, A.; Simões, E.A.F.; Yadav, P.D.; Chadha, M.S. Laboratory-Confirmed Avian Influenza A(H9N2) Virus Infection, India, 2019. Emerg. Infect. Dis. 2019, 25, 2328–2330. [Google Scholar] [CrossRef]
- Jallow, M.M.; Fall, A.; Barry, M.A.; Diop, B.; Sy, S.; Goudiaby, D.; Fall, M.; Enouf, V.; Niang, M.N.; Dia, N. Genetic Characterization of the First Detected Human Case of Low Pathogenic Avian Influenza A/H9N2 in Sub-Saharan Africa, Senegal. Emerg. Microbes Infect. 2020, 9, 1092–1095. [Google Scholar] [CrossRef]
- McCrone, J.T.; Woods, R.J.; Martin, E.T.; Malosh, R.E.; Monto, A.S.; Lauring, A.S. Stochastic Processes Constrain the within and between Host Evolution of Influenza Virus. eLife 2018, 7, e35962. [Google Scholar] [CrossRef]
- Zhang, Y.; Aevermann, B.D.; Anderson, T.K.; Burke, D.F.; Dauphin, G.; Gu, Z.; He, S.; Kumar, S.; Larsen, C.N.; Lee, A.J.; et al. Influenza Research Database: An Integrated Bioinformatics Resource for Influenza Virus Research. Nucleic Acids Res. 2017, 45, D466–D474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- WOAH Chapter 3.3.4 Avian Influenza (Including Infection with High Pathogenicity Avian Influenza Viruses). In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2022; World Organisation for Animal Health: Paris, France, 2022.
- Wan, H.; Perez, D.R. Amino Acid 226 in the Hemagglutinin of H9N2 Influenza Viruses Determines Cell Tropism and Replication in Human Airway Epithelial Cells. J. Virol. 2007, 81, 5181–5191. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Deng, G.; Song, J.; Tian, G.; Suo, Y.; Jiang, Y.; Guan, Y.; Bu, Z.; Kawaoka, Y.; Chen, H. Two Amino Acid Residues in the Matrix Protein M1 Contribute to the Virulence Difference of H5N1 Avian Influenza Viruses in Mice. Virology 2009, 384, 28–32. [Google Scholar] [CrossRef]
- Jiao, P.; Tian, G.; Li, Y.; Deng, G.; Jiang, Y.; Liu, C.; Liu, W.; Bu, Z.; Kawaoka, Y.; Chen, H. A Single-Amino-Acid Substitution in the NS1 Protein Changes the Pathogenicity of H5N1 Avian Influenza Viruses in Mice. J. Virol. 2008, 82, 1146–1154. [Google Scholar] [CrossRef]
- Zhu, W.; Zou, X.; Zhou, J.; Tang, J.; Shu, Y. Residues 41V and/or 210D in the NP Protein Enhance Polymerase Activities and Potential Replication of Novel Influenza (H7N9) Viruses at Low Temperature. Virol. J. 2015, 12, 71. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Yamada, S.; Fukuyama, S.; Murakami, S.; Zhao, D.; Uraki, R.; Watanabe, T.; Tomita, Y.; Macken, C.; Neumann, G.; et al. Virulence-Affecting Amino Acid Changes in the PA Protein of H7N9 Influenza A Viruses. J. Virol. 2014, 88, 3127–3134. [Google Scholar] [CrossRef]
- Elgendy, E.M.; Arai, Y.; Kawashita, N.; Daidoji, T.; Takagi, T.; Ibrahim, M.S.; Nakaya, T.; Watanabe, Y. Identification of Polymerase Gene Mutations That Affect Viral Replication in H5N1 Influenza Viruses Isolated from Pigeons. J. Gen. Virol. 2017, 98, 6–17. [Google Scholar] [CrossRef]
- Feng, X.; Wang, Z.; Shi, J.; Deng, G.; Kong, H.; Tao, S.; Li, C.; Liu, L.; Guan, Y.; Chen, H. Glycine at Position 622 in PB1 Contributes to the Virulence of H5N1 Avian Influenza Virus in Mice. J. Virol. 2016, 90, 1872–1879. [Google Scholar] [CrossRef]
- Graef, K.M.; Vreede, F.T.; Lau, Y.-F.; McCall, A.W.; Carr, S.M.; Subbarao, K.; Fodor, E. The PB2 Subunit of the Influenza Virus RNA Polymerase Affects Virulence by Interacting with the Mitochondrial Antiviral Signaling Protein and Inhibiting Expression of Beta Interferon. J. Virol. 2010, 84, 8433–8445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Hatta, M.; Watanabe, S.; Neumann, G.; Watanabe, T.; Kawaoka, Y. Role of Host-Specific Amino Acids in the Pathogenicity of Avian H5N1 Influenza Viruses in Mice. J. Gen. Virol. 2010, 91, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Zhang, Y.; Dong, L.; Wang, D.; Huang, W.; Xin, L.; Yang, L.; Zhao, X.; Li, Z.; Wang, W.; et al. A Comprehensive Surveillance of Adamantane Resistance among Human Influenza A Virus Isolated from Mainland China between 1956 and 2009. Antivir. Ther. 2010, 15, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Baba, K.; Inoue, K.; Okane, M.; Hata, S.; Shishido, T.; Naito, A.; Wildum, S.; Omoto, S. Comprehensive Assessment of Amino Acid Substitutions in the Trimeric RNA Polymerase Complex of Influenza A Virus Detected in Clinical Trials of Baloxavir Marboxil. Influenza Other Respir. Viruses 2021, 15, 389–395. [Google Scholar] [CrossRef]
- Tada, T.; Suzuki, K.; Sakurai, Y.; Kubo, M.; Okada, H.; Itoh, T.; Tsukamoto, K. NP Body Domain and PB2 Contribute to Increased Virulence of H5N1 Highly Pathogenic Avian Influenza Viruses in Chickens. J. Virol. 2011, 85, 1834–1846. [Google Scholar] [CrossRef] [PubMed]
- Wasilenko, J.L.; Sarmento, L.; Pantin-Jackwood, M.J. A Single Substitution in Amino Acid 184 of the NP Protein Alters the Replication and Pathogenicity of H5N1 Avian Influenza Viruses in Chickens. Arch. Virol. 2009, 154, 969–979. [Google Scholar] [CrossRef]
- Ayllon, J.; Domingues, P.; Rajsbaum, R.; Miorin, L.; Schmolke, M.; Hale, B.G.; Garcia-Sastre, A. A Single Amino Acid Substitution in the Novel H7N9 Influenza A Virus NS1 Protein Increases CPSF30 Binding and Virulence. J. Virol. 2014, 88, 12146–12151. [Google Scholar] [CrossRef] [PubMed]
- Kirunda, H.; Erima, B.; Tumushabe, A.; Kiconco, J.; Tugume, T.; Mulei, S.; Mimbe, D.; Mworozi, E.; Bwogi, J.; Luswa, L.; et al. Prevalence of Influenza A Viruses in Livestock and Free-Living Waterfowl in Uganda. BMC Vet. Res. 2014, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, L.; Shittu, I.; Fusaro, A.; Inuwa, B.; Zecchin, B.; Gado, D.; Schivo, A.; Bianco, A.; Laleye, A.; Gobbo, F.; et al. Live Bird Markets in Nigeria: A Potential Reservoir for H9N2 Avian Influenza Viruses. Viruses 2021, 13, 1445. [Google Scholar] [CrossRef]
- Youk, S.; Lee, D.-H.; Jeong, J.-H.; Pantin-Jackwood, M.J.; Song, C.; Swayne, D.E. Live Bird Markets as Evolutionary Epicentres of H9N2 Low Pathogenicity Avian Influenza Viruses in Korea. Emerg. Microbes Infect. 2020, 9, 616–627. [Google Scholar] [CrossRef]
- Elsayed, M.; Arafa, A.; Abdelwahab, S.; Hashish, A.; Youssef, A. Novel Reassortant of H9N2 Avian Influenza Viruses Isolated from Chickens and Quails in Egypt. Vet. World 2021, 14, 2142–2149. [Google Scholar] [CrossRef] [PubMed]
- Kandeil, A.; El-Shesheny, R.; Maatouq, A.; Moatasim, Y.; Cai, Z.; McKenzie, P.; Webby, R.; Kayali, G.; Ali, M.A. Novel Reassortant H9N2 Viruses in Pigeons and Evidence for Antigenic Diversity of H9N2 Viruses Isolated from Quails in Egypt. J. Gen. Virol. 2017, 98, 548–562. [Google Scholar] [CrossRef]
- Li, X.; Sun, J.; Lv, X.; Wang, Y.; Li, Y.; Li, M.; Liu, W.; Zhi, M.; Yang, X.; Fu, T.; et al. Novel Reassortant Avian Influenza A(H9N2) Virus Isolate in Migratory Waterfowl in Hubei Province, China. Front. Microbiol. 2020, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Suttie, A.; Karlsson, E.A.; Deng, Y.-M.; Horm, S.V.; Yann, S.; Tok, S.; Sorn, S.; Holl, D.; Tum, S.; Hurt, A.C.; et al. Influenza A(H5N1) Viruses with A(H9N2) Single Gene (Matrix or PB1) Reassortment Isolated from Cambodian Live Bird Markets. Virology 2018, 523, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, A.; Mahmoud, S.H.; Shehata, M.; Müller, C.; Kandeil, A.; El-Shesheny, R.; Nooh, H.Z.; Kayali, G.; Ali, M.A.; Pleschka, S. PA from a Recent H9N2 (G1-Like) Avian Influenza A Virus (AIV) Strain Carrying Lysine 367 Confers Altered Replication Efficiency and Pathogenicity to Contemporaneous H5N1 in Mammalian Systems. Viruses 2020, 12, 1046. [Google Scholar] [CrossRef]
- Mercan, Y.; Atim, G.; Kayed, A.E.; Azbazdar, M.E.; Kandeil, A.; Ali, M.A.; Rubrum, A.; McKenzie, P.; Webby, R.J.; Erima, B.; et al. Molecular Characterization of Closely Related H6N2 Avian Influenza Viruses Isolated from Turkey, Egypt, and Uganda. Viruses 2021, 13, 607. [Google Scholar] [CrossRef]
- Sun, Y.; Cong, Y.; Yu, H.; Ding, Z.; Cong, Y. Assessing the Effects of a Two-Amino Acid Flexibility in the Hemagglutinin 220-Loop Receptor-Binding Domain on the Fitness of Influenza A(H9N2) Viruses. Emerg. Microbes Infect. 2021, 10, 822–832. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, K.; Li, B.; Chen, Y.; Qiu, Z.; Xing, J.; Huang, J.; Hu, C.; Huang, Y.; Li, H.; et al. A Risk Marker of Tribasic Hemagglutinin Cleavage Site in Influenza A (H9N2) Virus. Commun. Biol. 2021, 4, 71. [Google Scholar] [CrossRef]
- Li, Y.; Liu, M.; Sun, Q.; Zhang, H.; Zhang, H.; Jiang, S.; Liu, S.; Huang, Y. Genotypic Evolution and Epidemiological Characteristics of H9N2 Influenza Virus in Shandong Province, China. Poult. Sci. 2019, 98, 3488–3495. [Google Scholar] [CrossRef]
- Sealy, J.E.; Yaqub, T.; Peacock, T.P.; Chang, P.; Ermetal, B.; Clements, A.; Sadeyen, J.-R.; Mehboob, A.; Shelton, H.; Bryant, J.E.; et al. Association of Increased Receptor-Binding Avidity of Influenza A(H9N2) Viruses with Escape from Antibody-Based Immunity and Enhanced Zoonotic Potential. Emerg. Infect. Dis. 2018, 25, 63–72. [Google Scholar] [CrossRef]
- Sorrell, E.M.; Wan, H.; Araya, Y.; Song, H.; Perez, D.R. Minimal Molecular Constraints for Respiratory Droplet Transmission of an Avian-Human H9N2 Influenza A Virus. Proc. Natl. Acad. Sci. USA 2009, 106, 7565–7570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Qian, J.; Song, Y.; Ming, D. The Adaptability of H9N2 Avian Influenza A Virus to Humans: A Comparative Docking Simulation Study. Biochem. Biophys. Res. Commun. 2020, 529, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Kawashita, N.; Ibrahim, M.S.; Elgendy, E.M.; Daidoji, T.; Ono, T.; Takagi, T.; Nakaya, T.; Matsumoto, K.; Watanabe, Y. PB2 Mutations Arising during H9N2 Influenza Evolution in the Middle East Confer Enhanced Replication and Growth in Mammals. PLoS Pathog. 2019, 15, e1007919. [Google Scholar] [CrossRef]
- Sun, X.; Belser, J.A.; Maines, T.R. Adaptation of H9N2 Influenza Viruses to Mammalian Hosts: A Review of Molecular Markers. Viruses 2020, 12, 541. [Google Scholar] [CrossRef]
- Chen, Z.; Huang, Q.; Yang, S.; Su, S.; Li, B.; Cui, N.; Xu, C. A Well-Defined H9N2 Avian Influenza Virus Genotype with High Adaption in Mammals Was Prevalent in Chinese Poultry Between 2016 to 2019. Viruses 2020, 12, 432. [Google Scholar] [CrossRef]
- Li, J.; Ishaq, M.; Prudence, M.; Xi, X.; Hu, T.; Liu, Q.; Guo, D. Single Mutation at the Amino Acid Position 627 of PB2 That Leads to Increased Virulence of an H5N1 Avian Influenza Virus during Adaptation in Mice Can Be Compensated by Multiple Mutations at Other Sites of PB2. Virus Res. 2009, 144, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Kariithi, H.M.; Welch, C.N.; Ferreira, H.L.; Pusch, E.A.; Ateya, L.O.; Binepal, Y.S.; Apopo, A.A.; Dulu, T.D.; Afonso, C.L.; Suarez, D.L. Genetic Characterization and Pathogenesis of the First H9N2 Low Pathogenic Avian Influenza Viruses Isolated from Chickens in Kenyan Live Bird Markets. Infect. Genet. Evol. 2020, 78, 104074. [Google Scholar] [CrossRef]
- Arbi, M.; Souiai, O.; Rego, N.; Larbi, I.; Naya, H.; Ghram, A.; Houimel, M. Historical Origins and Zoonotic Potential of Avian Influenza Virus H9N2 in Tunisia Revealed by Bayesian Analysis and Molecular Characterization. Arch. Virol. 2020, 165, 1527–1540. [Google Scholar] [CrossRef]
- Barberis, A.; Boudaoud, A.; Gorrill, A.; Loupias, J.; Ghram, A.; Lachheb, J.; Alloui, N.; Ducatez, M.F. Full-Length Genome Sequences of the First H9N2 Avian Influenza Viruses Isolated in the Northeast of Algeria. Virol. J. 2020, 17, 108. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Sequence Features | Phenotype | Frequency |
|---|---|---|---|
| HA | T197, G520 | Acquisition of respiratory droplet transmission and human-like clinical symptoms | All |
| Q226L | Increased virus binding to α2–6, enhanced replication in mammalian cells and ferrets, enhanced contact transmission in ferrets | ||
| M1 M2 | N30D, I43M, T215A | Increased virulence in mice | S31N absent in 1 |
| S31N | Increased Amantadine resistance | ||
| NP | M105V, A184K | Increased virulence in chicken [38,39] | All |
| E210D | Increased polymerase activity in mammalian cell line | ||
| NS | P42S | Increased virulence and decreased antiviral response in mice | All |
| I106M | Increased viral replication in mammalian cells and virulence in mice [40] | ||
| PA | S37A, N383D | Increased polymerase activity in mammalian and avian cell line | All E199D in 8 |
| E199D | Decreased sensitivity to Baloxivir | ||
| PB1 | D3V | Increased polymerase activity and replication in mammalian and avian cell lines | All |
| D622G | Increased polymerase activity in mammalian cell lines | ||
| PB2 | L89V, G309D, T339K, R477G, I495V, A676V | Increased polymerase activity in mammalian cell line and increased virulence in mice | All |
| D9N | Increased virulence in mice | ||
| K256R | Increased polymerase activity in mammalian cell lines |
| Clade | Oman/2747 | Bd/0994 | HK/1073 | HK/33982 | qa/Bd/19462 | ck/Benin | ck/Bd/46240 | ck/BD/46129 | Collection Date | Passage History | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference antigen | ||||||||||||
| 1 | A/Oman/2747/2019 IDCDC-RG66A | G1 | 2560 | 320 | < | 40 | 640 | 320 | 640 | 640 | V1E2/E1 | |
| 2 | A/Bangladesh/0994/2011-IDCDC-RG31 | G1 | 1280 | 1280 | < | 80 | 640 | 640 | 1280 | 2560 | V1E2/E1 | |
| 3 | A/Hong Kong/1073/99 | G1 | < | < | 160 | 640 | < | < | < | < | C4 | |
| 4 | A/Hong Kong/33982/2009-PR8-IDCDC-RG-26 | G1 | < | < | 160 | 1280 | < | < | < | < | V1E3/E2/E1 | |
| 5 | A/quail/Bangladesh/19462/2013 | G1 | 320 | 40 | 10 | 80 | 320 | 80 | 160 | 160 | E2 | |
| 6 | A/chicken/Benin/19-A-01-145-E/2019 | G1 | 1280 | 320 | < | 80 | 640 | 640 | 640 | 1280 | E1/E2 | |
| 7 | A/chicken/Bangladesh/46240/2020 | G1 | 80 | 80 | < | 40 | 80 | 160 | 640 | 1280 | E1 | |
| 8 | A/chicken/Bangladesh/46129/2020 | G1 | 640 | 640 | < | 80 | 640 | 320 | 1280 | 2560 | E1 | |
| Test antigens | ||||||||||||
| 9 | A/chicken/MUWRP-UGANDA/944/2019 | G1 | 1280 | 2560 | < | 20 | 640 | 320 | 1280 | 2560 | 03/13/19 | X/E1 |
| 10 | A/chicken/MUWRP-UGANDA/951/2019 | G1 | 2560 | 2560 | < | 40 | 1280 | 640 | 1280 | 2560 | 03/13/19 | X/E1 |
| 11 | A/chicken/MUWRP-UGANDA/957/2019 | G1 | 1280 | 1280 | < | 20 | 320 | 320 | 1280 | 2560 | 04/24/19 | X/E1 |
| 12 | A/chicken/MUWRP-UGANDA/977/2019 | G1 | 1280 | 1280 | < | 20 | 640 | 320 | 1280 | 2560 | 06/19/19 | X/E1 |
| 13 | A/chicken/MUWRP-UGANDA/1038/2019 | G1 | 640 | 1280 | < | 20 | 320 | 320 | 640 | 1280 | 09/25/19 | X/E1 |
| 14 | A/chicken/MUWRP-UGANDA/1064/2019 | G1 | 1280 | 1280 | 10 | 20 | 640 | 320 | 1280 | 2560 | 11/20/19 | X/E1 |
| 15 | A/chicken/MUWRP-UGANDA/1082/2019 | G1 | 1280 | 1280 | 10 | 10 | 640 | 320 | 2560 | 2560 | 12/19/19 | X/E1 |
| Serum production: P = prime | CDC | P | P | P | P | P | P | P | ||||
 .
.Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atim, G.; Tugume, T.; Ukuli, Q.A.; Erima, B.; Mubiru, A.; Kibuuka, H.; Mworozi, E.; McKenzie, P.; Turner, J.C.M.; Walker, D.; et al. Genetic Evolution of Avian Influenza A (H9N2) Viruses Isolated from Domestic Poultry in Uganda Reveals Evidence of Mammalian Host Adaptation, Increased Virulence and Reduced Sensitivity to Baloxavir. Viruses 2022, 14, 2074. https://doi.org/10.3390/v14092074
Atim G, Tugume T, Ukuli QA, Erima B, Mubiru A, Kibuuka H, Mworozi E, McKenzie P, Turner JCM, Walker D, et al. Genetic Evolution of Avian Influenza A (H9N2) Viruses Isolated from Domestic Poultry in Uganda Reveals Evidence of Mammalian Host Adaptation, Increased Virulence and Reduced Sensitivity to Baloxavir. Viruses. 2022; 14(9):2074. https://doi.org/10.3390/v14092074
Chicago/Turabian StyleAtim, Gladys, Titus Tugume, Qouilazoni A. Ukuli, Bernard Erima, Andrew Mubiru, Hannah Kibuuka, Edison Mworozi, Pamela McKenzie, Jasmine C. M. Turner, David Walker, and et al. 2022. "Genetic Evolution of Avian Influenza A (H9N2) Viruses Isolated from Domestic Poultry in Uganda Reveals Evidence of Mammalian Host Adaptation, Increased Virulence and Reduced Sensitivity to Baloxavir" Viruses 14, no. 9: 2074. https://doi.org/10.3390/v14092074