
Complete Genome and Molecular Characterization of a New Cyprinid Herpesvirus 2 (CyHV-2) SH-01 Strain Isolated from Cultured Crucian Carp
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of Virus and DNA Extraction
2.2. DNA Sequencing, Genome Assembly, and Annotation
2.3. Analysis of the Genome Structure and Molecular Characterization of CyHV-2 SH-01
2.4. Comparison of Genomic Structure and Evolutionary Relationships among SH-01 and the Other Seven Strains
3. Results
3.1. Genome Structure and Composition
3.2. Chronological Characteristics of Gene Expression
3.3. Features of the Predicted Functional Protein Encoded by SH-01
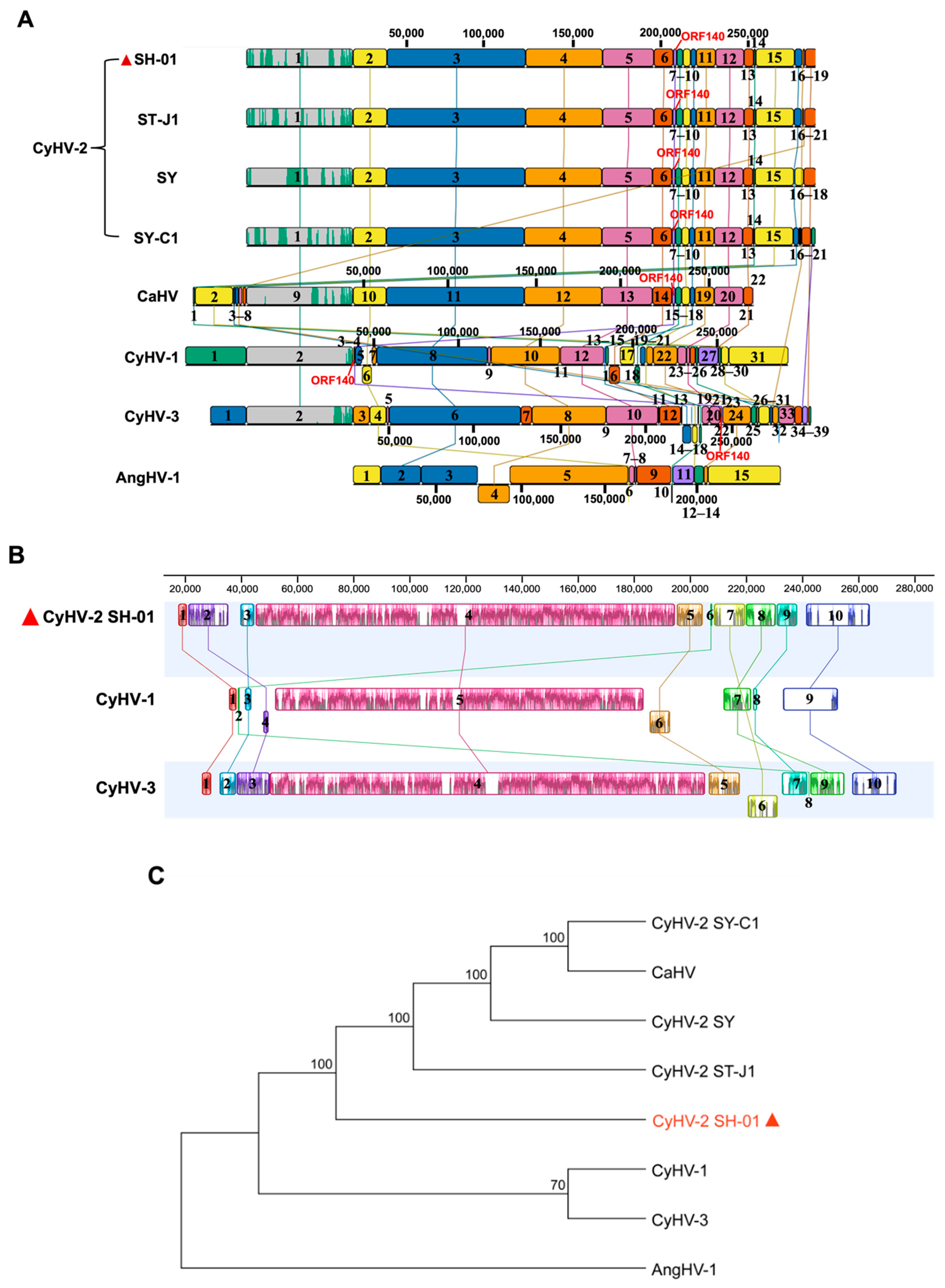
3.4. Comparison of Genome Structure and ORFs Arrangement
3.5. Genomic Evolutionary Relationships among SH-01 and the Other Seven Strains
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jung, S.J.; Miyazaki, T. Herpesviral haematopoietic necrosis of goldfish, Carassius auratus (L.). J. Fish Dis. 1995, 18, 211–220. [Google Scholar] [CrossRef]
- Stephens, F.J.; Raidal, S.R.; Jones, B. Haematopoietic necrosis in a goldfish (Carassius auratus) associated with an agent morphologically similar to herpesvirus. Aust. Vet. J. 2004, 82, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Groff, J.M.; LaPatra, S.E.; Munn, R.J.; Zinkl, J.G. A viral epizootic in cultured populations of juvenile goldfish due to a putative herpesvirus etiology. J. Vet. Diagn. Investig. 1998, 10, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, A.E.; Khoo, L.; LaPatra, S.E.; Bonar, C.; Key, D.W.; Garner, M.; Lee, M.V.; Hanson, L. Goldfish hematopoietic necrosis herpesvirus (cyprinid herpesvirus 2) in the USA: Molecular confirmation of isolates from diseased fish. J. Aquat. Anim. Health 2006, 18, 11–18. [Google Scholar] [CrossRef]
- Jeffery, K.R.; Bateman, K.; Bayley, A.; Feist, S.W.; Hulland, J.; Longshaw, C.; Stone, D.; Woolford, G.; Way, K. Isolation of a cyprinid herpesvirus 2 from goldfish, Carassius auratus (L.), in the UK. J. Fish Dis. 2007, 30, 649–656. [Google Scholar] [CrossRef]
- Doszpoly, A.; Maria, B.A.; György, C.; Ádám, D.; Mária, L.; Balázs, H. Introduction of the family Alloherpesviridae: The first molecular detection of herpesviruses of cyprinid fish in Hungary. Magy. Allatorv. Lapja 2011, 133, 174–181. [Google Scholar]
- Daněk, T.; Kalous, L.; Vesel, T.; Krásová, E.; Reschová, S.; Rylková, K.; Kulich, P.; Petrtýl, M.; Pokorová, D.; Knytl, M. Massive mortality of Prussian carp Carassius gibelio in the upper Elbe basin associated with herpesviral hematopoietic necrosis (CyHV-2). Dis. Aquat. Org. 2012, 102, 87–95. [Google Scholar] [CrossRef]
- Wang, L.; He, J.; Liang, L.; Zheng, X.; Jia, P.; Shi, X.; Lan, W.; Xie, J.; Liu, H.; Xu, P. Mass mortality caused by Cyprinid Herpesvirus 2 (CyHV-2) in Prussian carp (Carassius gibelio) in China. Bull. Eur. Assoc. Fish Pathol. 2012, 32, 164–173. [Google Scholar]
- Fichi, G.; Cardeti, G.; Cocumelli, C.; Vendramin, N.; Toffan, A.; Eleni, C.; Siemoni, N.; Fischetti, R.; Susini, F. Detection of Cyprinid herpesvirus 2 in association with an Aeromonas sobria infection of Carassius carassius (L.), in Italy. J. Fish Dis. 2013, 36, 823–830. [Google Scholar] [CrossRef]
- Boitard, P.-M.; Baud, M.; Labrut, S.; de Boisséson, C.; Jamin, M.; Bigarré, L. First detection of Cyprinid Herpesvirus 2 (CyHV-2) in goldfish (Carassius auratus) in France. J. Fish Dis. 2016, 39, 673–680. [Google Scholar] [CrossRef]
- Haenen, O.L.M.; Way, K.; Gorgoglione, B.; Ito, T.; Paley, R.; Bigarré, L.; Waltzek, T. Novel viral infections threatening Cyprinid fish. Bull. Eur. Assoc. Fish Pathol. 2016, 36, 11–23. [Google Scholar]
- Sahoo, P.K.; Swaminathan, T.R.; Abraham, T.J.; Kumar, R.; Pattanayak, S.; Mohapatra, A.; Rath, S.S.; Patra, A.; Adikesavalu, H.; Sood, N.; et al. Detection of goldfish haematopoietic necrosis herpes virus (Cyprinid herpesvirus-2) with multi-drug resistant Aeromonas hydrophila infection in goldfish: First evidence of any viral disease outbreak in ornamental freshwater aquaculture farms in India. Acta Trop. 2016, 161, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, S.; Bergmann, S.M.; Keeling, C.; Lany, C.; Schütze, H.; Schmidt-Posthaus, H. Herpesviral Hematopoietic Necrosis in Goldfish in Switzerland: Early Lesions in Clinically Normal Goldfish (Carassius auratus). Vet. Pathol. 2016, 53, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Adamek, M.; Hellmann, J.; Jung-Schroers, V.; Teitge, F.; Steinhagen, D. CyHV-2 transmission in traded goldfish stocks in Germany—A case study. J. Fish Dis. 2018, 41, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Kalayci, G.; Ozkan, B.; Pekmez, K.; Kaplan, M.; Mefut, A.; Anil Cagirgan, A. First detection of Cyprinid herpesvirus-2 (CyHV-2) followed by screening and monitoring studies in Goldfish (Carassius Auratus) in Turkey. Bull. Eur. Assoc. Fish Pathol. 2018, 38, 94–103. [Google Scholar]
- Panicz, R.; Sadowski, J.; Eljasik, P. Detection of Cyprinid herpesvirus 2 (CyHV-2) in symptomatic ornamental types of goldfish (Carassius auratus) and asymptomatic common carp (Cyprinus carpio) reared in warm-water cage culture. Aquaculture 2019, 504, 131–138. [Google Scholar] [CrossRef]
- Adler, B.; Sattler, C.; Adler, H. Herpesviruses and Their Host Cells: A Successful Liaison. Trends Microbiol. 2017, 25, 229–241. [Google Scholar] [CrossRef]
- Collins-McMillen, D.; Buehler, J.; Peppenelli, M.; Goodrum, F. Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation. Viruses 2018, 10, 444. [Google Scholar] [CrossRef]
- Eide, K.E.; Miller-Morgan, T.; Heidel, J.R.; Kent, M.L.; Bildfell, R.J.; Lapatra, S.; Watson, G.; Jin, L. Investigation of koi herpesvirus latency in koi. J. Virol. 2011, 85, 4954–4962. [Google Scholar] [CrossRef]
- Wei, C.; Iida, H.; Chuah, Q.; Tanaka, M.; Kato, G.; Sano, M. Persistence of cyprinid herpesvirus 2 in asymptomatic goldfish Carassius auratus (L.) that survived an experimental infection. J. Fish Dis. 2019, 42, 913–921. [Google Scholar] [CrossRef]
- Wei, C.; Kakazu, T.; Chuah, Q.Y.; Tanaka, M.; Kato, G.; Sano, M. Reactivation of cyprinid herpesvirus 2 (CyHV-2) in asymptomatic surviving goldfish Carassius auratus (L.) under immunosuppression. Fish Shellfish Immunol. 2020, 103, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Waltzek, T.B.; Kelley, G.O.; Stone, D.M.; Way, K.; Hanson, L.; Fukuda, H.; Hirono, I.; Aoki, T.; Davison, A.J.; Hedrick, R.P. Koi herpesvirus represents a third cyprinid herpesvirus (CyHV-3) in the family Herpesviridae. J. Gen. Virol. 2005, 86, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Waltzek, T.B.; Kelley, G.O.; Alfaro, M.E.; Kurobe, T.; Davison, A.J.; Hedrick, R.P. Phylogenetic relationships in the family Alloherpesviridae. Dis. Aquat. Organ. 2009, 84, 179–194. [Google Scholar] [CrossRef] [PubMed]
- van Beurden, S.J.; Bossers, A.; Voorbergen-Laarman, M.H.A.; Haenen, O.L.M.; Peters, S.; Abma-Henkens, M.H.C.; Peeters, B.P.H.; Rottier, P.J.M.; Engelsma, M.Y. Complete genome sequence and taxonomic position of anguillid herpesvirus 1. J. Gen. Virol. 2010, 91, 880–887. [Google Scholar] [CrossRef]
- Doszpoly, A.; Kovács, E.R.; Bovo, G.; LaPatra, S.E.; Harrach, B.; Benko, M. Molecular confirmation of a new herpesvirus from catfish (Ameiurus melas) by testing the performance of a novel PCR method, designed to target the DNA polymerase gene of alloherpesviruses. Arch. Virol. 2008, 153, 2123–2127. [Google Scholar] [CrossRef]
- Zeng, X.-T.; Chen, Z.-Y.; Deng, Y.-S.; Gui, J.-F.; Zhang, Q.-Y. Complete genome sequence and architecture of crucian carp Carassius auratus herpesvirus (CaHV). Arch. Virol. 2016, 161, 3577–3581. [Google Scholar] [CrossRef]
- Wen, J.; Xu, Y.; Su, M.; Lu, L.; Wang, H. Susceptibility of Goldfish to Cyprinid Herpesvirus 2 (CyHV-2) SH01 Isolated from Cultured Crucian Carp. Viruses 2021, 13, 1761. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar]
- Davison, A.J.; Kurobe, T.; Gatherer, D.; Cunningham, C.; Korf, I.; Fukuda, H.; Hedrick, R.P.; Waltzek, T.B. Comparative genomics of carp herpesviruses. J. Virol. 2013, 87, 2908–2922. [Google Scholar] [CrossRef]
- Yuan, Y. Identification and characterization of herpesviral immediate-early genes. Methods Mol. Biol. 2005, 292, 231–244. [Google Scholar]
- Gruffat, H.; Marchione, R.; Manet, E. Herpesvirus late gene expression: A viral-specific pre-initiation complex is key. Front. Microbiol. 2016, 7, 869. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Lu, L.; Wang, B.; Yu, J.; Wang, H. Identification of the Immediate-Early Genes of Cyprinid Herpesvirus 2. Viruses 2020, 12, 994. [Google Scholar] [CrossRef]
- Naom, I.S.; Morton, S.J.; Leach, D.R.F.; Lloyd, R.G. Molecular organization of sbcC, a gene that affects genetic recombination and the viability of DNA palindromes in Escherichia coli K-12. Nucleic Acids Res. 1989, 17, 8033–8045. [Google Scholar] [CrossRef]
- Zhang, J.; Ge, W.; Zhan, P.; De Clercq, E.; Liu, X. Retroviral restriction factors TRIM5α: Therapeutic strategy to inhibit HIV-1 replication. Curr. Med. Chem. 2011, 18, 2649–2654. [Google Scholar] [CrossRef]
- Maris, C.; Dominguez, C.; Allain, F.H.-T. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005, 272, 2118–2131. [Google Scholar] [CrossRef]
- Bailey-Elkin, B.A.; van Kasteren, P.B.; Snijder, E.J.; Kikkert, M.; Mark, B.L. Viral OTU deubiquitinases: A structural and functional comparison. PLoS Pathog. 2014, 10, e1003894. [Google Scholar] [CrossRef]
- Rajcani, J.; Szenthe, K.; Banati, F.; Szathmary, S. Survey of Epstein Barr virus (EBV) immunogenic proteins and their epitopes: Implications for vaccine preparation. Recent Pat. Anti-Infect. Drug Discov. 2014, 9, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.L.; Williams, L.R.; White, C.; Forrest, C.; Zuo, J.; Rowe, M. The Missing Link in Epstein-Barr Virus Immune Evasion: The BDLF3 Gene Induces Ubiquitination and Downregulation of Major Histocompatibility Complex Class I (MHC-I) and MHC-II. J. Virol. 2015, 90, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Bates, P.; Rivera-Gonzalez, R.; Gu, B.; DeLuca, N.A. ICP4, the major transcriptional regulatory protein of herpes simplex virus type 1, forms a tripartite complex with TATA-binding protein and TFIIB. J. Virol. 1993, 67, 4676–4687. [Google Scholar] [CrossRef]
- Li, L.; Luo, Y.; Gao, Z.; Huang, J.; Zheng, X.; Nie, H.; Zhang, J.; Lin, L.; Yuan, J. Molecular characterisation and prevalence of a new genotype of Cyprinid herpesvirus 2 in mainland China. Can. J. Microbiol. 2015, 61, 381–387. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, Y.; Li, K.; Hu, X.; Wang, C.; Cao, G.; Xue, R.; Gong, C. The complete genome of Cyprinid herpesvirus 2, a new strain isolated from Allogynogenetic crucian carp. Virus Res. 2018, 256, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Hirono, I.; Kurokawa, K.; Fukuda, H.; Nahary, R.; Eldar, A.; Davison, A.J.; Waltzek, T.B.; Bercovier, H.; Hedrick, R.P. Genome sequences of three koi herpesvirus isolates representing the expanding distribution of an emerging disease threatening koi and common carp worldwide. J. Virol. 2007, 81, 5058–5065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.E.; Treangen, T.J.; Messeguer, X.; Perna, N.T. Analyzing patterns of microbial evolution using the mauve genome alignment system. Methods Mol. Biol. 2007, 396, 135–152. [Google Scholar]
- McGeoch, D.J. Molecular evolution of large DNA viruses of eukaryotes. Semin. Virol. 1992, 3, 399–409. [Google Scholar]
- Nishiyama, Y. Herpes simplex virus gene products: The accessories reflect her lifestyle well. Rev. Med. Virol. 2004, 14, 33–46. [Google Scholar] [CrossRef]
- Isomura, H.; Stinski, M.F. Coordination of late gene transcription of human cytomegalovirus with viral DNA synthesis: Recombinant viruses as potential therapeutic vaccine candidates. Expert Opin. Ther. Targets 2013, 17, 157–166. [Google Scholar] [CrossRef]
- Boyne, J.R.; Whitehouse, A. γ-2 Herpes virus post-transcriptional gene regulation. Clin. Microbiol. Infect. 2006, 12, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Lumangtad, L.A.; Bell, T.W. The signal peptide as a new target for drug design. Bioorg. Med. Chem. Lett. 2020, 30, 127115. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ning, S. New Look of EBV LMP1 Signaling Landscape. Cancers 2021, 13, 5451. [Google Scholar] [CrossRef] [PubMed]
- Patané, J.S.L.; Martins, J., Jr.; Rangel, L.T.; Belasque, J.; Digiampietri, L.A.; Facincani, A.P.; Ferreira, R.M.; Jaciani, F.J.; Zhang, Y.; Varani, A.M.; et al. Origin and diversification of Xanthomonas citri subsp. citri pathotypes revealed by inclusive phylogenomic, dating, and biogeographic analyses. BMC Genom. 2019, 20, 700. [Google Scholar] [CrossRef]
- Yancopoulos, S.; Attie, O.; Friedberg, R. Efficient sorting of genomic permutations by translocation, inversion and block interchange. Bioinformatics 2005, 21, 3340–3346. [Google Scholar] [CrossRef]
- Lunter, G.; Ponting, C.P.; Hein, J. Genome-wide identification of human functional DNA using a neutral indel model. PLoS Comput. Biol. 2006, 2, e5. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Virus | Size (bp) | Nucleotide Composition (%) | No. of ORFs | Identity (%) k | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genome | U g | TR h | G+C | Genome i | Unique j | U g | TR h | |||
| CyHV-2 | SH-01 | 290,428 | 260,586 | 14,921 | 51.60 | 154 | 150 | 146 | 4 | *** |
| ST-J1 a | 290,304 | 260,238 | 15,033 | 51.70 | 154 | 150 | 146 | 4 | 99.60 | |
| SY-C1 b | 289,365 | 259,555 | 14,905 | 51.60 | 143 | 140 | 137 | 3 | 98.53 | |
| SY c | 290,455 | 259,749 | 15,353 | 51.60 | 154 | 150 | 146 | 4 | 98.35 | |
| CyHV-1 a | 291,144 | 224,784 | 33,180 | 51.30 | 143 | 137 | 131 | 6 | 42.72 | |
| CyHV-3 d | 295,146 | 250,208 | 22,469 | 59.20 | 163 | 155 | 147 | 8 | 44.54 | |
| AngHV-1 e | 248,526 | 227,258 | 10,634 | 53.00 | 134 | 129 | 124 | 5 | 36.52 | |
| CaHV f | 275,348 | - | - | 51.73 | 150 | 150 | - | - | 92.63 | |
| SH-01 | CyHV-1 | Change (bp) b | CyHV-3 | Change (bp) b | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LCB | Position | Length (bp) | LCB | Position | Length (bp) | LCB | Position | Length (bp) | ||
| LCB1 | 17,215–20,413 | 3199 | LCB1 | 35,374–38,196 | 2823 | −376 | LCB1 | 25,754–29,079 | 3326 | +127 |
| LCB2 | 20,806–35,267 | 14,462 | LCB4 | 47,773–49,683 | 1911 | −12,551 | LCB3 | 38,157–49,904 | 11,748 | −2714 |
| LCB3 | 39,346–44,586 | 5241 | LCB3 | 41,045–43,535 | 2491 | −2750 | LCB2 | 32,129–37,868 | 5740 | +499 |
| LCB4 | 44,914–195,079 | 150,166 | LCB5 | 51,890–183,835 | 131,946 | −18,220 | LCB4 | 50,081–205,795 | 155,715 | +5549 |
| LCB5 | 195,702–204,934 | 9233 | LCB6 | 185,864–193,284 | 7421 | −1812 | LCB5 | 207,192–218,326 | 11,135 | +1902 |
| LCB6 | 207,694–208,337 | 644 | LCB2 | 38,663–39,134 | 472 | −172 | LCB8 | 242,836–243,294 | 459 | −185 |
| LCB7 | 209,037–220,265 | 11,229 | none a | none a | none a | −11,229 | LCB6 | 221,009–231,789 | 10,781 | −448 |
| LCB8 | 220,500–231,255 | 10,756 | LCB7 | 212,442–222,389 | 9948 | –808 | LCB9 | 243,510–255,637 | 12,128 | +1372 |
| LCB9 | 231,395–238,734 | 7340 | LCB8 | 222,900–224,492 | 1593 | −5747 | LCB7 | 233,309–242,408 | 9100 | +1760 |
| LCB10 | 241,885–264,893 | 23,009 | LCB9 | 233,688–253,332 | 19,645 | −3364 | LCB10 | 258,414–274,200 | 15,787 | −7222 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Wen, J.; Xiao, S.; Wei, C.; Yu, F.; Roengjit, P.; Lu, L.; Wang, H. Complete Genome and Molecular Characterization of a New Cyprinid Herpesvirus 2 (CyHV-2) SH-01 Strain Isolated from Cultured Crucian Carp. Viruses 2022, 14, 2068. https://doi.org/10.3390/v14092068
Yang J, Wen J, Xiao S, Wei C, Yu F, Roengjit P, Lu L, Wang H. Complete Genome and Molecular Characterization of a New Cyprinid Herpesvirus 2 (CyHV-2) SH-01 Strain Isolated from Cultured Crucian Carp. Viruses. 2022; 14(9):2068. https://doi.org/10.3390/v14092068
Chicago/Turabian StyleYang, Jia, Jinxuan Wen, Simin Xiao, Chang Wei, Fei Yu, Patarida Roengjit, Liqun Lu, and Hao Wang. 2022. "Complete Genome and Molecular Characterization of a New Cyprinid Herpesvirus 2 (CyHV-2) SH-01 Strain Isolated from Cultured Crucian Carp" Viruses 14, no. 9: 2068. https://doi.org/10.3390/v14092068