
FDA-Approved Inhibitors of RTK/Raf Signaling Potently Impair Multiple Steps of In Vitro and Ex Vivo Influenza A Virus Infections
 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Human Precision-Cut Lung Slices (hPCLSs)
2.3. Inhibitors
2.4. In Vitro and Ex Vivo Cytotoxicity Assays
2.5. Virus Infections
2.6. Immunofluorescent Staining and Imaging
2.7. Polymerase Activity Assay
2.8. Viral Entry Assay and Confocal Microscopy
2.9. SMKI Resistance Analysis
2.10. Viral Egress Assay and qPCR Analysis
2.11. Statistical Analyses
3. Results
3.1. Identification of SMKIs That Potently Inhibit In Vitro IAV Infections
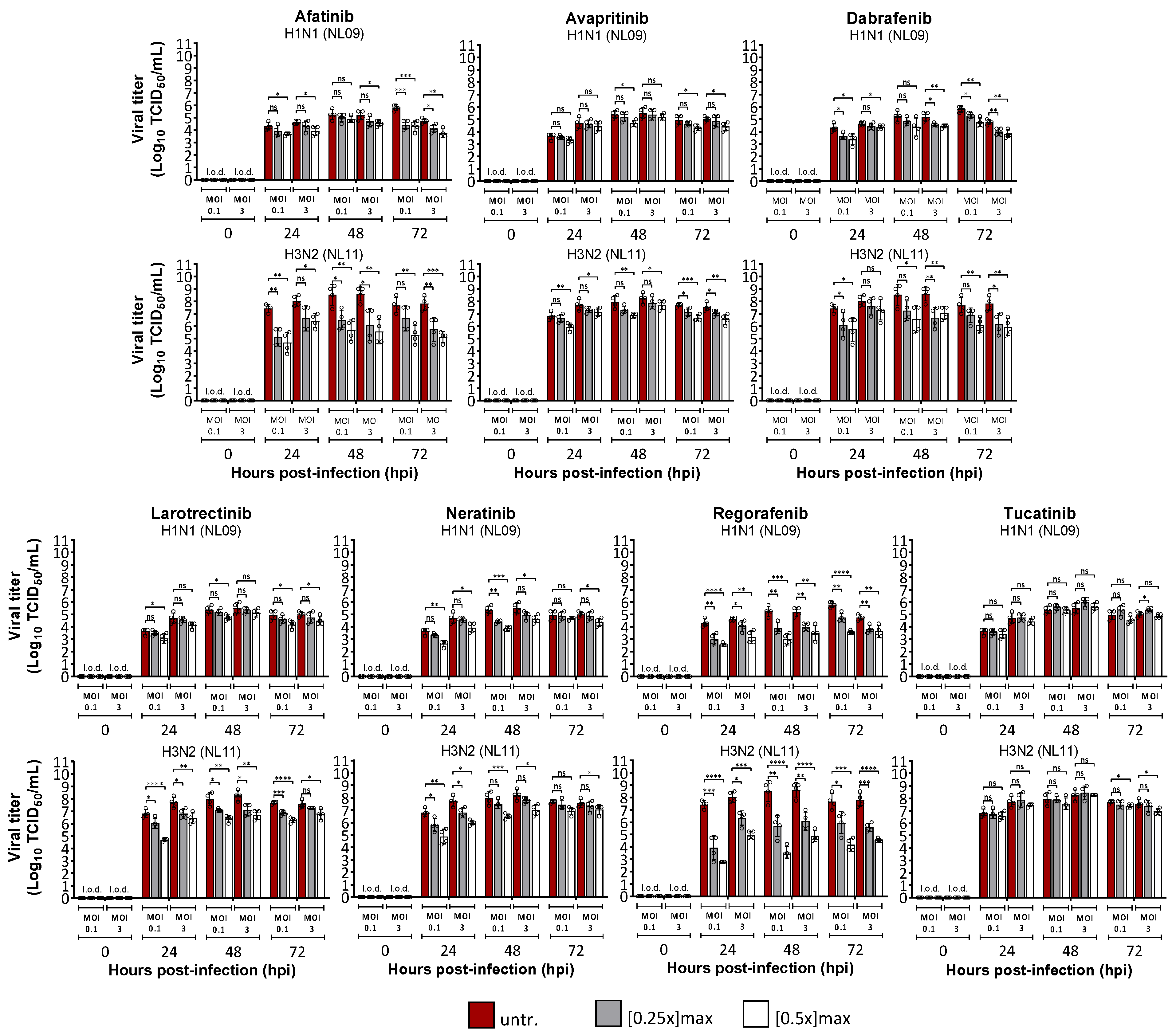
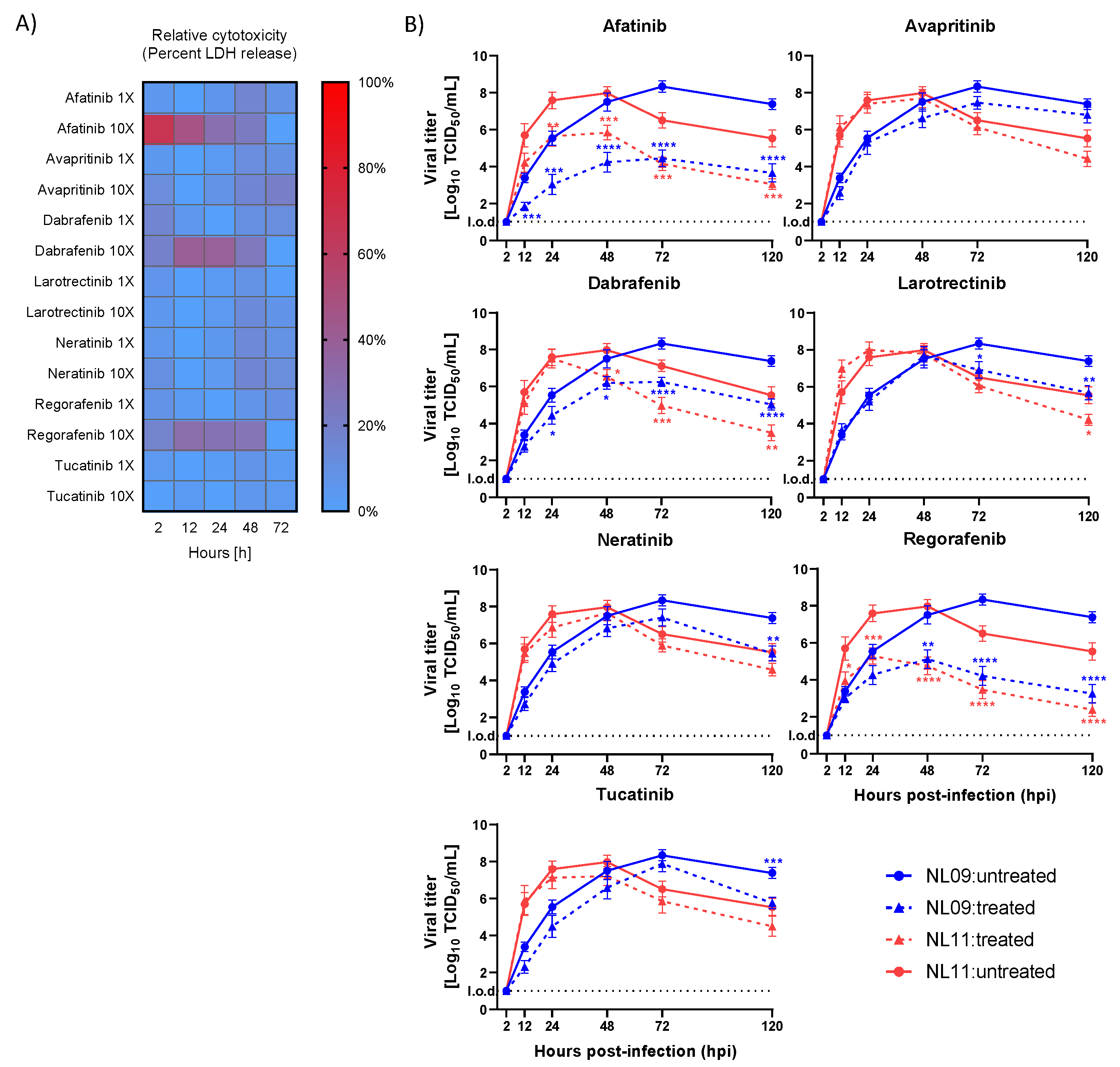
3.2. Effects of SMKI Treatment on In Vitro Cell Viability, Infectivity and Viral Spread
3.3. SMKIs Differentially Inhibit Various Steps of IAV Infection Cycle
3.4. Select SMKIs Exhibit Potent Antiviral Activity in IAV-Infected hPCLSs
3.5. Tested SMKIs Have a High Genetic Barrier of Resistance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Influenza (Seasonal). Fact Sheet. November 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 11 January 2022).
- Kuiken, T.; Riteau, B.; Fouchier, R.; Rimmelzwaan, G. Pathogenesis of influenza virus infections: The good, the bad and the ugly. Curr. Opin. Virol. 2012, 2, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Kroeze, E.J.B.V.; Fouchier, R.A.M.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, Y.-H.; Yang, Z.-Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016, 13, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Nair, H.; Brooks, W.A.; Katz, M.; Roca, A.; Berkley, J.A.; Madhi, S.A.; Simmerman, J.M.; Gordon, A.; Sato, M.; Howie, S.; et al. Global burden of respiratory infections due to seasonal influenza in young children: A systematic review and meta-analysis. Lancet 2011, 378, 1917–1930. [Google Scholar] [CrossRef]
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, L.; Cox, N.; Anderson, L.J.; Fukuda, K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 2003, 289, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.G.; Govorkova, E. Continuing challenges in influenza. Ann. N. Y. Acad. Sci. 2014, 1323, 115–139. [Google Scholar] [CrossRef]
- Monto, A.S.; Fukuda, K. Lessons from influenza pandemics of the last 100 years. Clin. Infect. Dis. 2020, 70, 951–957. [Google Scholar] [CrossRef]
- Nelson, M.I.; Simonsen, L.; Viboud, C.; Miller, M.A.; Holmes, E. The origin and global emergence of adamantane resistant A/H3N2 influenza viruses. Virology 2009, 388, 270–278. [Google Scholar] [CrossRef]
- Lackenby, A.; Besselaar, T.G.; Daniels, R.S.; Fry, A.; Gregory, V.; Gubareva, L.V.; Huang, W.; Hurt, A.C.; Leang, S.-K.; Lee, R.T.C.; et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors and status of novel antivirals, 2016–2017. Antivir. Res. 2018, 157, 38–46. [Google Scholar] [CrossRef]
- Nitsch-Osuch, A.; Brydak, L.B. Influenza viruses resistant to neuraminidase inhibitors. Acta Biochim. Pol. 2014, 61, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Omoto, S.; Speranzini, V.; Hashimoto, T.; Noshi, T.; Yamaguchi, H.; Kawai, M.; Kawaguchi, K.; Uehara, T.; Shishido, T.; Naito, A.; et al. Characterization of influenza virus variants induced by treatment with the endonuclease inhibitor baloxavir marboxil. Sci. Rep. 2018, 8, 9633. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Hayden, F.G.; Kawaguchi, K.; Omoto, S.; Hurt, A.C.; De Jong, M.D.; Hirotsu, N.; Sugaya, N.; Lee, N.; Baba, K.; et al. Treatment-emergent influenza variant viruses with reduced baloxavir susceptibility: Impact on clinical and virologic outcomes in uncomplicated influenza. J. Infect. Dis. 2020, 221, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 2017, 17, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Sharma, N.R.; Ly, H.; Parslow, T.G.; Liang, Y. Receptor tyrosine kinase inhibitors that block replication of influenza a and other viruses. Antimicrob. Agents Chemother. 2011, 55, 5553–5559. [Google Scholar] [CrossRef]
- Kurokawa, M.; Ochiai, H.; Nakajima, K.; Niwayama, S. Inhibitory effect of protein kinase C inhibitor on the replication of influenza type A virus. J. Gen. Virol. 1990, 71, 2149–2155. [Google Scholar] [CrossRef]
- Pleschka, S.; Wolff, T.; Ehrhardt, C.; Hobom, G.; Planz, O.; Rapp, U.R.; Ludwig, S. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 2001, 3, 301–305. [Google Scholar] [CrossRef]
- Kumar, N.; Liang, Y.; Parslow, T.G.; Liang, Y. Receptor tyrosine kinase inhibitors block multiple steps of influenza A virus replication. J. Virol. 2011, 85, 2818–2827. [Google Scholar] [CrossRef]
- Elbahesh, H.; Bergmann, S.; Russell, C.J. Focal adhesion kinase (FAK) regulates polymerase activity of multiple influenza A virus subtypes. Virology 2016, 499, 369–374. [Google Scholar] [CrossRef]
- Elbahesh, H.; Cline, T.; Baranovich, T.; Govorkova, E.A.; Schultz-Cherry, S.; Russell, C.J. Novel roles of focal adhesion kinase in cytoplasmic entry and replication of influenza A viruses. J. Virol. 2014, 88, 6714–6728. [Google Scholar] [CrossRef]
- Meineke, R.; Rimmelzwaan, G.F.; Elbahesh, H. Influenza virus infections and cellular kinases. Viruses 2019, 11, 171. [Google Scholar] [CrossRef] [Green Version]
- Meineke, R.; Stelz, S.; Busch, M.; Werlein, C.; Kühnel, M.; Jonigk, D.; Rimmelzwaan, G.F.; Elbahesh, H. Ex vivo validation of six FDA-approved non-receptor tyrosine kinase inhibitors (NRTKIs) as antivirals to pandemic and seasonal influenza A viruses. bioRxiv 2022. [CrossRef]
- Liu, G.; Betts, C.; Cunoosamy, D.M.; Åberg, P.M.; Hornberg, J.J.; Sivars, K.B.; Cohen, T.S. Use of precision cut lung slices as a translational model for the study of lung biology. Respir. Res. 2019, 20, 162. [Google Scholar] [CrossRef] [PubMed]
- Preuß, E.B.; Schubert, S.; Werlein, C.; Stark, H.; Braubach, P.; Höfer, A.; Plucinski, E.K.; Shah, H.R.; Geffers, R.; Sewald, K.; et al. The challenge of long-term cultivation of human precision-cut lung slices. Am. J. Pathol. 2021, 192, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Viana, F.; O’Kane, C.M.; Schroeder, G.N. Precision-cut lung slices: A powerful ex vivo model to investigate respiratory infectious diseases. Mol. Microbiol. 2021, 117, 578–588. [Google Scholar] [CrossRef]
- Kirchhoff, J.; Uhlenbruck, S.; Keil, G.M.; Schwegmann-Wessels, C.; Ganter, M.; Herrler, G. Infection of differentiated airway epithelial cells from caprine lungs by viruses of the bovine respiratory disease complex. Veter Microbiol. 2014, 170, 58–64. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Grishagin, I.V. Automatic cell counting with image. J. Anal. Biochem. 2015, 473, 63–65. [Google Scholar] [CrossRef]
- Azzeh, M.; Flick, R.; Hobom, G. Functional analysis of the influenza A virus cRNA promoter and construction of an ambisense transcription system. Virology 2001, 289, 400–410. [Google Scholar] [CrossRef]
- Deng, T.; Sharps, J.L.; Brownlee, G.G. Role of the influenza virus heterotrimeric RNA polymerase complex in the initiation of replication. J. Gen. Virol. 2006, 87, 3373–3377. [Google Scholar] [CrossRef]
- Hoffmann, H.-H.; Palese, P.; Shaw, M.L. Modulation of influenza virus replication by alteration of sodium ion transport and protein kinase C activity. Antivir. Res. 2008, 80, 124–134. [Google Scholar] [CrossRef]
- World Health Organization. WHO Information for the Molecular Detection of Influenza Viruses. Available online: www.who.int/influenza/gisrs_laboratory/Protocols_influenza_virus_detection_Nov_2018.pdf (accessed on 18 January 2022).
- Siegers, J.Y.; van de Bildt, M.W.G.; Lin, Z.; Leijten, L.M.; Lavrijssen, R.A.M.; Bestebroer, T.; Spronken, M.I.J.; De Zeeuw, C.I.; Gao, Z.; Schrauwen, E.J.A.; et al. Viral factors important for efficient replication of influenza A viruses in cells of the central nervous system. J. Virol. 2019, 93, e02273-18. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, V.; Danov, O.; Konzok, S.; Obernolte, H.; Dehmel, S.; Braubach, P.; Jonigk, D.; Fieguth, H.-G.; Zardo, P.; Warnecke, G.; et al. Assessment of the cytotoxic and immunomodulatory effects of substances in human precision-cut lung slices. J. Vis. Exp. 2018, 135, 57042. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Radtke, K.; Dohner, K.; Sodeik, B. Viral interactions with the cytoskeleton: A hitchhiker’s guide to the cell. Cell. Microbiol. 2006, 8, 387–400. [Google Scholar] [CrossRef]
- Zheng, K.; Kitazato, K.; Wang, Y. Viruses exploit the function of epidermal growth factor receptor. Rev. Med. Virol. 2014, 24, 274–286. [Google Scholar] [CrossRef]
- Schreiber, A.; Boff, L.; Anhlan, D.; Krischuns, T.; Brunotte, L.; Schuberth, C.; Wedlich-Söldner, R.; Drexler, H.; Ludwig, S. Dissecting the mechanism of signaling-triggered nuclear export of newly synthesized influenza virus ribonucleoprotein complexes. Proc. Natl. Acad. Sci. USA 2020, 117, 16557–16566. [Google Scholar] [CrossRef]
- Shin, Y.-K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Effect of the phosphatidylinositol 3-kinase/Akt pathway on influenza A virus propagation. J. Gen. Virol. 2007, 88, 942–950. [Google Scholar] [CrossRef]
- Ehrhardt, C. From virus entry to release: The diverse functions of PI3K during RNA virus infections. Futur. Virol. 2011, 6, 1225–1239. [Google Scholar] [CrossRef]
- Arrese, M.; Portela, A. Serine 3 is critical for phosphorylation at the N-terminal end of the nucleoprotein of influenza virus A/Victoria/3/75. J. Virol. 1996, 70, 3385–3391. [Google Scholar] [CrossRef] [Green Version]
- Hsiang, T.-Y.; Zhou, L.; Krug, R.M. Roles of the Phosphorylation of Specific Serines and Threonines in the NS1 Protein of Human Influenza A Viruses. J. Virol. 2012, 86, 10370–10376. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S. Targeting cell signalling pathways to fight the flu: Towards a paradigm change in anti-influenza therapy. J. Antimicrob. Chemother. 2009, 64, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Marjuki, H.; Gornitzky, A.; Marathe, B.M.; Ilyushina, N.A.; Aldridge, J.R.; Desai, G.; Webby, R.J.; Webster, R.G. Influenza A virus-induced early activation of ERK and PI3K mediates V-ATPase-dependent intracellular pH change required for fusion. Cell. Microbiol. 2010, 13, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Planz, O. Development of cellular signaling pathway inhibitors as new antivirals against influenza. Antivir. Res. 2013, 98, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, Z.; Bi, Y.; Sun, L.; Liu, X.; Liu, W. Tyrosine 132 phosphorylation of influenza A virus m1 protein is crucial for virus replication by controlling the nuclear import of M1. J. Virol. 2013, 87, 6182–6191. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, S.; Hu, Y.; Li, D.; Cui, J.; Xue, J.; Zhang, G.; Khachigian, L.M.; Wong, J.; Sun, L.; et al. Regulatory roles of c-jun in H5N1 influenza virus replication and host inflammation. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 2479–2488. [Google Scholar] [CrossRef]
- Turrell, L.; Hutchinson, E.C.; Vreede, F.T.; Fodor, E. Regulation of influenza A virus nucleoprotein oligomerization by phosphorylation. J. Virol. 2014, 89, 1452–1455. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; Denham, E.M.; Thomas, B.; Trudgian, D.C.; Hester, S.S.; Ridlova, G.; York, A.; Turrell, L.; Fodor, E. Mapping the phosphoproteome of influenza a and b viruses by mass spectrometry. PLoS Pathog. 2012, 8, e1002993. [Google Scholar] [CrossRef]
- York, A.; Hutchinson, E.C.; Fodor, E. Interactome analysis of the influenza A virus transcription/replication machinery identifies protein phosphatase 6 as a cellular factor required for efficient virus replication. J. Virol. 2014, 88, 13284–13299. [Google Scholar] [CrossRef]
- Eierhoff, T.; Hrincius, E.R.; Rescher, U.; Ludwig, S.; Ehrhardt, C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog. 2010, 6, e1001099. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, H.D.; Eisfeld, A.J.; Stratton, K.G.; Heller, N.C.; Bramer, L.M.; Wen, J.; McDermott, J.E.; Gralinski, L.E.; Sims, A.C.; Le, M.Q.; et al. The role of EGFR in influenza pathogenicity: Multiple network-based approaches to identify a key regulator of non-lethal infections. Front. Cell Dev. Biol. 2019, 7, 200. [Google Scholar] [CrossRef] [PubMed]
- Ueki, I.F.; Min-Oo, G.; Kalinowski, A.; Ballon-Landa, E.; Lanier, L.L.; Nadel, J.A.; Koff, J.L. Respiratory virus–induced EGFR activation suppresses IRF1-dependent interferon λ and antiviral defense in airway epithelium. J. Exp. Med. 2013, 210, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Pan, W.; Wang, S.; Pan, C.; Ning, H.; Huang, S.; Chiu, S.-H.; Chen, J.-L. Protein tyrosine phosphatase SHP2 suppresses host innate immunity against influenza A virus by regulating EGFR-mediated signaling. J. Virol. 2021, 95, e02001–e02020. [Google Scholar] [CrossRef]
- Rheault, T.R.; Stellwagen, J.C.; Adjabeng, G.M.; Hornberger, K.R.; Petrov, K.G.; Waterson, A.G.; Dickerson, S.H.; Mook, R.A., Jr.; Laquerre, S.G.; King, A.J.; et al. Discovery of dabrafenib: A selective inhibitor of raf kinases with antitumor activity against B-raf-driven tumors. ACS Med. Chem. Lett. 2013, 4, 358–362. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.-H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2010, 129, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Lesch, M.; Luckner, M.; Meyer, M.; Weege, F.; Gravenstein, I.; Raftery, M.; Sieben, C.; Martin-Sancho, L.; Imai-Matsushima, A.; Welke, R.-W.; et al. RNAi-based small molecule repositioning reveals clinically approved urea-based kinase inhibitors as broadly active antivirals. PLoS Pathog. 2019, 15, e1007601. [Google Scholar] [CrossRef]
- Holzberg, M.; Boergeling, Y.; Schräder, T.; Ludwig, S.; Ehrhardt, C. Vemurafenib limits influenza A virus propagation by targeting multiple signaling pathways. Front. Microbiol. 2017, 8, 2426. [Google Scholar] [CrossRef]
- Chen, P.-H.; Chen, X.; He, X. Platelet-derived growth factors and their receptors: Structural and functional perspectives. Biochim. Biophys. Acta 2013, 1834, 2176–2186. [Google Scholar] [CrossRef]
- Lee, I.-T.; Yang, C.-M. Inflammatory signalings involved in airway and pulmonary diseases. Mediat. Inflamm. 2013, 2013, 791231. [Google Scholar] [CrossRef]
- Vrijens, P.; Noppen, S.; Boogaerts, T.; Vanstreels, E.; Ronca, R.; Chiodelli, P.; Laporte, M.; Vanderlinden, E.; Liekens, S.; Stevaert, A.; et al. Influenza virus entry via the GM3 ganglioside-mediated platelet-derived growth factor receptor β signalling pathway. J. Gen. Virol. 2019, 100, 583–601. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Ivy, S.P.; Wick, J.Y.; Kaufman, B.M. An overview of small-molecule inhibitors of VEGFR signaling. Nat. Rev. Clin. Oncol. 2009, 6, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Liong, S.; Oseghale, O.; To, E.E.; Brassington, K.; Erlich, J.R.; Luong, R.; Liong, F.; Brooks, R.; Martin, C.; O’Toole, S.; et al. Influenza A virus causes maternal and fetal pathology via innate and adaptive vascular inflammation in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 24964–24973. [Google Scholar] [CrossRef]
- Pacheco-Hernández, L.M.; Ramírez-Noyola, J.A.; Gómez-García, I.A.; Ignacio-Cortés, S.; Zúñiga, J.; Choreño-Parra, J.A. Comparing the cytokine storms of COVID-19 and pandemic influenza. J. Interf. Cytokine Res. 2022, 42, 369–392. [Google Scholar] [CrossRef]
- An, Q.; Han, C.; Zhou, Y.; Li, F.; Li, D.; Zhang, X.; Yu, Z.; Duan, Z.; Kan, Q. Matrine induces cell cycle arrest and apoptosis with recovery of the expression of miR-126 in the A549 non-small cell lung cancer cell line. Mol. Med. Rep. 2016, 14, 4042–4048. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves first new flu drug in 20 years. Nat. Rev. Drug Discov. 2018, 17, 853. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Sugaya, N.; Hirotsu, N.; Lee, N.; De Jong, M.D.; Hurt, A.C.; Ishida, T.; Sekino, H.; Yamada, K.; Portsmouth, S.; et al. Baloxavir Marboxil for Uncomplicated Influenza in Adults and Adolescents. N. Engl. J. Med. 2018, 379, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.L.; Amornphimoltham, P.; Schmidt, M.; Wilson, P.A.; Gutkind, J.S.; Chiorini, J.A. Epidermal growth factor receptor is a co-receptor for adeno-associated virus serotype 6. Nat. Med. 2010, 16, 662–664. [Google Scholar] [CrossRef]
- Peng, X.; Zhou, Y.; Tao, Y.; Liu, S. Nasopharyngeal carcinoma: The role of the EGFR in epstein–barr virus infection. Pathogens 2021, 10, 1113. [Google Scholar] [CrossRef]
- Iwamoto, M.; Saso, W.; Nishioka, K.; Ohashi, H.; Sugiyama, R.; Ryo, A.; Ohki, M.; Yun, J.-H.; Park, S.-Y.; Ohshima, T.; et al. The machinery for endocytosis of epidermal growth factor receptor coordinates the transport of incoming hepatitis B virus to the endosomal network. J. Biol. Chem. 2019, 295, 800–807. [Google Scholar] [CrossRef]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-J.; Min, C.-K.; Hancock, M.; Streblow, D.N.; Caposio, P.; Goodrum, F.D.; Yurochko, A.D. Human cytomegalovirus host interactions: EGFR and host cell signaling is a point of convergence between viral infection and functional changes in infected cells. Front. Microbiol. 2021, 12, 660901. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Sterk, R.T.; DeHaro, S.A.; Ozbun, M.A. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J. Virol. 2013, 87, 2508–2517. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Xiang, Y.; Wang, X.; Wang, Q.; Zhong, M.; Wang, S.; Wang, X.; Fan, J.; Kitazato, K.; Wang, Y. Epidermal growth factor receptor-PI3K signaling controls cofilin activity to facilitate herpes simplex virus 1 entry into neuronal cells. mBio 2014, 5, e00958-13. [Google Scholar] [CrossRef]
- Lee, S.; Currier, M.G.; Hotard, A.L.; Meng, J.; Pretto, C.; Power, U.F.; Villenave, R.; Shields, M.D.; Chi, M.H.; Peebles, R.S.; et al. Epidermal growth factor receptor (EGFR) mediates cell fusion and infectivity of respiratory syncytial virus (RSV). J. Allergy Clin. Immunol. 2014, 133, Ab71. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, S.; Shen, Y.; Yang, Q. Epidermal growth factor receptor is a co-factor for transmissible gastroenteritis virus entry. Virology 2018, 521, 33–43. [Google Scholar] [CrossRef]
- Beerli, C.; Yakimovich, A.; Kilcher, S.; Reynoso, G.V.; Fläschner, G.; Muller, D.J.; Hickman, H.; Mercer, J. Vaccinia virus hijacks EGFR signalling to enhance virus spread through rapid and directed infected cell motility. Nat. Microbiol. 2018, 4, 216–225. [Google Scholar] [CrossRef]
- Pasquale, G.D.; Davidson, B.L.; Stein, C.S.; Martins, I.; Scudiero, D.; Monks, A.; Chiorini, J.A. Identification of PDGFR as a receptor for AAV-5 transduction. Nat. Med. 2003, 9, 1306–1312. [Google Scholar] [CrossRef]
- Wu, Y.; Prager, A.; Boos, S.; Resch, M.; Brizić, I.; Mach, M.; Wildner, S.; Scrivano, L.; Adler, B. Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-α as a key for entry. PLoS Pathog. 2017, 13, e1006281. [Google Scholar] [CrossRef]
- Liu, X.; Cohen, J.I. The role of PI3K/Akt in human herpesvirus infection: From the bench to the bedside. Virology 2015, 479–480, 568–577. [Google Scholar] [CrossRef] [Green Version]
- Campadelli-Fiume, G.; Collins-McMillen, D.; Gianni, T.; Yurochko, A.D. Integrins as herpesvirus receptors and mediators of the host signalosome. Annu. Rev. Virol. 2016, 3, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Alkharsah, K.R. VEGF upregulation in viral infections and its possible therapeutic implications. Int. J. Mol. Sci. 2018, 19, 1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meineke, R.; Stelz, S.; Busch, M.; Werlein, C.; Kühnel, M.; Jonigk, D.; Rimmelzwaan, G.F.; Elbahesh, H. FDA-Approved Inhibitors of RTK/Raf Signaling Potently Impair Multiple Steps of In Vitro and Ex Vivo Influenza A Virus Infections. Viruses 2022, 14, 2058. https://doi.org/10.3390/v14092058
Meineke R, Stelz S, Busch M, Werlein C, Kühnel M, Jonigk D, Rimmelzwaan GF, Elbahesh H. FDA-Approved Inhibitors of RTK/Raf Signaling Potently Impair Multiple Steps of In Vitro and Ex Vivo Influenza A Virus Infections. Viruses. 2022; 14(9):2058. https://doi.org/10.3390/v14092058
Chicago/Turabian StyleMeineke, Robert, Sonja Stelz, Maximilian Busch, Christopher Werlein, Mark Kühnel, Danny Jonigk, Guus F. Rimmelzwaan, and Husni Elbahesh. 2022. "FDA-Approved Inhibitors of RTK/Raf Signaling Potently Impair Multiple Steps of In Vitro and Ex Vivo Influenza A Virus Infections" Viruses 14, no. 9: 2058. https://doi.org/10.3390/v14092058