
Bovine Herpesvirus-1 Glycoprotein M Mediates the Translocation to the Golgi Apparatus and Packaging of VP8
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Plasmids
2.3. Antibodies
2.4. Transfection
2.5. Preparation of Cell Lysates
2.6. Immunoprecipitation and Western Blotting
2.7. Construction of Recombinant Virus
2.8. Viral Growth Kinetics
2.9. Confocal Microscopy
2.10. Transmission Electron Microscopy (TEM)
3. Results
3.1. Glycoprotein M Interacts with VP8
3.2. Glycoprotein M and VP8 Colocalize at the Golgi Apparatus
3.3. A Domain between Amino Acids 538–632 in VP8 Interacts with a Domain between Amino Acids 210–300 in gM
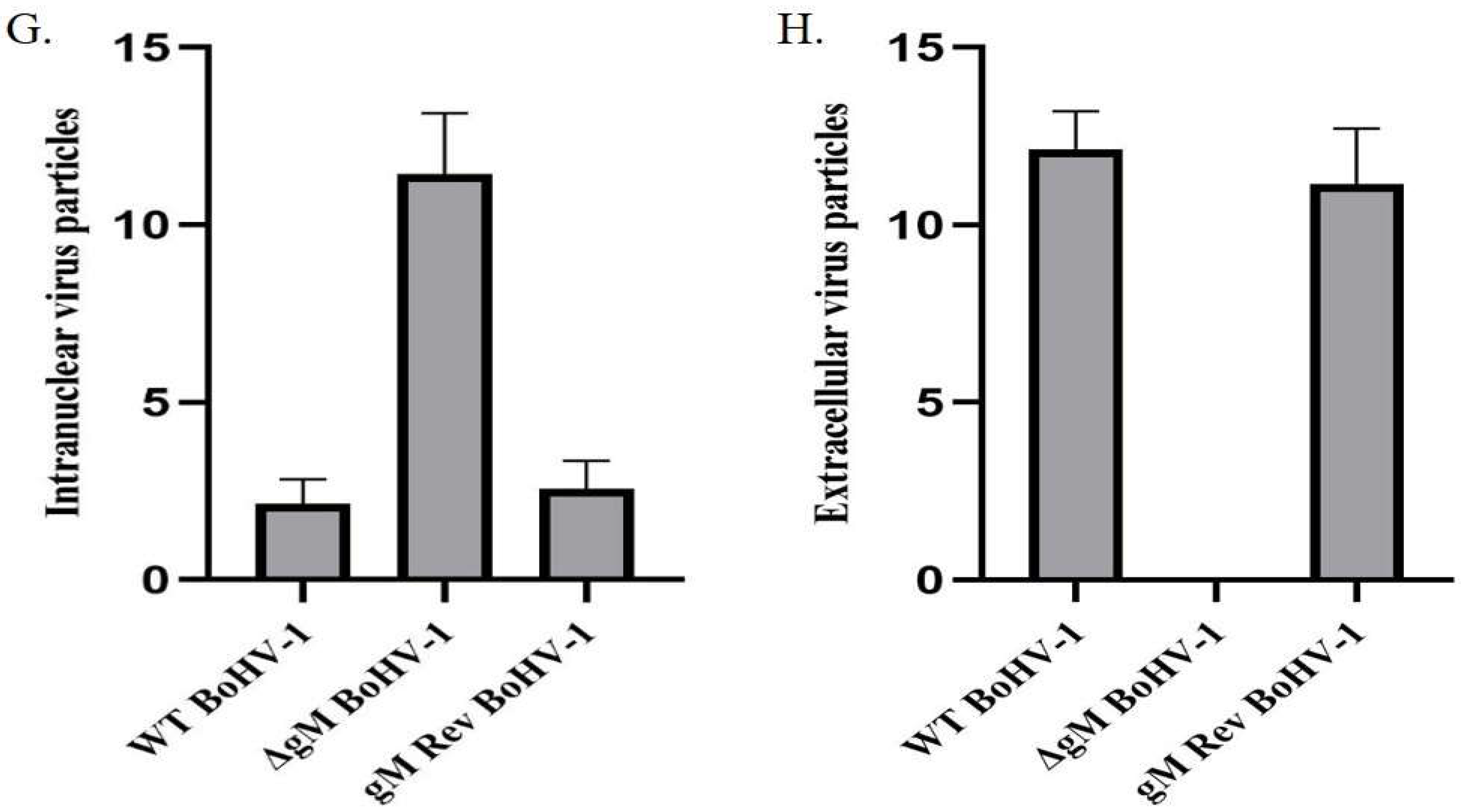
3.4. The ΔgM BoHV-1 Titer Is Significantly Reduced and Its Egress Is Affected
3.5. ΔgM BoHV-1 Shows Delayed Egress Compared to WT BoHV-1 and ΔgM Rev BoHV-1
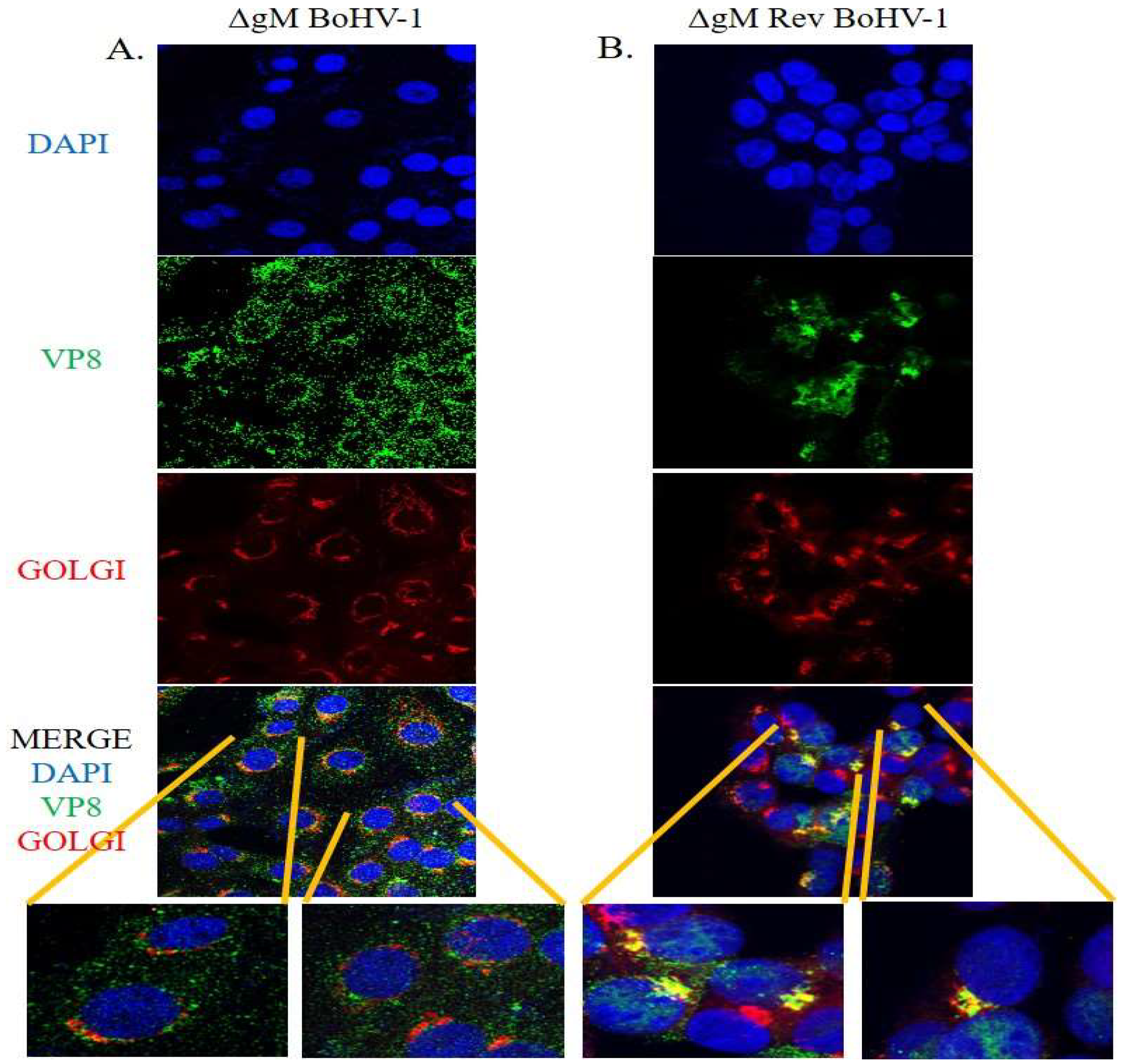
3.6. VP8 Does Not Localize to the Golgi in the Absence of gM
3.7. The Amount of VP8 in Mature Virus Is Considerably Reduced in gM-Deleted Virus
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levings, R.L.; Roth, J.A. Immunity to bovine herpesvirus 1: I. Viral lifecycle and innate immunity. Anim. Health Res. Rev. 2013, 14, 88–102. [Google Scholar] [CrossRef]
- Lucchese, L.; Benkirane, A.; Hakimi, I.; El Idrissi, A.; Natale, A. Seroprevalence study of the main causes of abortion in dairy cattle in Morocco. Vet. Ital. 2016, 52, 13–19. [Google Scholar] [CrossRef]
- Haanes, E.J.; Thomsen, D.R.; Martin, S.; Homa, F.L.; Lowery, D.E. The bovine herpesvirus 1 maturational proteinase and scaffold proteins can substitute for the homologous herpes simplex virus type 1 proteins in the formation of hybrid type B capsids. J. Virol. 1995, 69, 7375–7379. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, V.A.; Maher-Sturgess, S.L.; Snider, M.G.; Lawman, Z.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. A UL47 gene deletion mutant of bovine herpesvirus type 1 exhibits impaired growth in cell culture and lack of virulence in cattle. J. Virol. 2010, 84, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.J.; Fraefel, C.; Cunningham, A.L.; Diefenbach, R.J. Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res. 2009, 145, 173–186. [Google Scholar] [CrossRef] [PubMed]
- van Drunen Littel-van den Hurk, S.; Garzon, S.; van den Hurk, J.V.; Babiuk, L.A.; Tijssen, P. The role of the major tegument protein VP8 of bovine herpesvirus-1 in infection and immunity. Virology 1995, 206, 413–425. [Google Scholar] [CrossRef]
- Afroz, S.; Garg, R.; Fodje, M.; van Drunen Littel-van den Hurk, S. The Major Tegument Protein of Bovine Herpesvirus 1, VP8, Interacts with DNA Damage Response Proteins and Induces Apoptosis. J. Virol. 2018, 92, e00773-18. [Google Scholar] [CrossRef]
- Zhang, K.; Brownlie, R.; Snider, M.; van Drunen Littel-van den Hurk, S. Phosphorylation of Bovine Herpesvirus 1 VP8 Plays a Role in Viral DNA Encapsidation and Is Essential for Its Cytoplasmic Localization and Optimal Virion Incorporation. J. Virol. 2016, 90, 4427–4440. [Google Scholar] [CrossRef]
- Zheng, C.; Brownlie, R.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. Characterization of nuclear localization and export signals of the major tegument protein VP8 of bovine herpesvirus-1. Virology 2004, 324, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Afroz, S.; Brownlie, R.; Snider, M.; van Drunen Littel-van den Hurk, S. Regulation and function of phosphorylation on VP8, the major tegument protein of bovine herpesvirus 1. J. Virol. 2015, 89, 4598–4611. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Donovan, T.; Sucharita, S.; Brownlie, R.; Snider, M.; Tikoo, S.K.; van Drunen Littel-van den Hurk, S. US3 Kinase-Mediated Phosphorylation of Tegument Protein VP8 Plays a Critical Role in the Cellular Localization of VP8 and Its Effect on the Lipid Metabolism of Bovine Herpesvirus 1-Infected Cells. J. Virol. 2019, 93, e02151-18. [Google Scholar] [CrossRef] [PubMed]
- Labiuk, S.L.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. Major tegument protein VP8 of bovine herpesvirus 1 is phosphorylated by viral US3 and cellular CK2 protein kinases. J. Gen. Virol. 2009, 90 Pt 12, 2829–2839. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; van Drunen Littel-van den Hurk, S.; Babiuk, L.A.; Liang, X. Characterization of cell-binding properties of bovine herpesvirus 1 glycoproteins B, C, and D: Identification of a dual cell-binding function of gB. J. Virol. 1995, 69, 4758–4768. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.L.; Israel, B.A.; Letchworth, G.J., 3rd. Monoclonal antibody analysis of bovine herpesvirus-1 glycoprotein antigenic areas relevant to natural infection. Virology 1988, 165, 338–347. [Google Scholar] [CrossRef]
- Dawson, N.; Sillitoe, I.; Marsden, R.L.; Orengo, C.A. The Classification of Protein Domains. In Bioinformatics: Volume I: Data, Sequence Analysis, and Evolution; Keith, J.M., Ed.; Springer: New York, NY, USA, 2017; pp. 137–164. [Google Scholar] [CrossRef]
- Sucharita, S.; Zhang, K.; van Drunen Littel-van den Hurk, S. VP8, the Major Tegument Protein of Bovine Herpesvirus-1, Is Partially Packaged during Early Tegument Formation in a VP22-Dependent Manner. Viruses 2021, 13, 1854. [Google Scholar] [CrossRef]
- Mettenleiter, T.C. Intriguing interplay between viral proteins during herpesvirus assembly or: The herpesvirus assembly puzzle. Vet. Microbiol. 2006, 113, 163–169. [Google Scholar] [CrossRef]
- Maringer, K.; Stylianou, J.; Elliott, G. A network of protein interactions around the herpes simplex virus tegument protein VP22. J. Virol. 2012, 86, 12971–12982. [Google Scholar] [CrossRef]
- Crump, C. Virus Assembly and Egress of HSV. Adv. Exp. Med. Biol. 2018, 1045, 23–44. [Google Scholar] [CrossRef]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument Assembly and Secondary Envelopment of Alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef] [Green Version]
- McGeoch, D.J.; Dalrymple, M.A.; Davison, A.J.; Dolan, A.; Frame, M.C.; McNab, D.; Perry, L.J.; Scott, J.E.; Taylor, P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 1988, 69 Pt 7, 1531–1574. [Google Scholar] [CrossRef]
- Dolan, A.; Jamieson, F.E.; Cunningham, C.; Barnett, B.C.; McGeoch, D.J. The genome sequence of herpes simplex virus type 2. J. Virol. 1998, 72, 2010–2021. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.X.; Zhu, X.P.; Letchworth, G.J. Bovine herpesvirus 1 glycoprotein M forms a disulfide-linked heterodimer with the U(L)49.5 protein. J. Virol. 1998, 72, 3029–3036. [Google Scholar] [CrossRef] [PubMed]
- Brack, A.R.; Dijkstra, J.M.; Granzow, H.; Klupp, B.G.; Mettenleiter, T.C. Inhibition of virion maturation by simultaneous deletion of glycoproteins E, I, and M of pseudorabies virus. J. Virol. 1999, 73, 5364–5372. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Scott, J.E. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 1986, 67 Pt 9, 1759–1816. [Google Scholar] [CrossRef] [PubMed]
- Telford, E.A.; Watson, M.S.; McBride, K.; Davison, A.J. The DNA sequence of equine herpesvirus-1. Virology 1992, 189, 304–316. [Google Scholar] [CrossRef]
- El Kasmi, I.; Lippé, R. Herpes simplex virus 1 gN partners with gM to modulate the viral fusion machinery. J. Virol. 2015, 89, 2313–2323. [Google Scholar] [CrossRef]
- Graul, M.; Kisielnicka, E.; Rychłowski, M.; Verweij, M.C.; Tobler, K.; Ackermann, M.; Wiertz, E.; Bieńkowska-Szewczyk, K.; Lipińska, A.D. Transmembrane regions of bovine herpesvirus 1-encoded UL49.5 and glycoprotein M regulate complex maturation and ER-Golgi trafficking. J. Gen. Virol. 2019, 100, 497–510. [Google Scholar] [CrossRef]
- Striebinger, H.; Funk, C.; Raschbichler, V.; Bailer, S.M. Subcellular Trafficking and Functional Relationship of the HSV-1 Glycoproteins N and M. Viruses 2016, 8, 83. [Google Scholar] [CrossRef]
- Dijkstra, J.M.; Gerdts, V.; Klupp, B.G.; Mettenleiter, T.C. Deletion of glycoprotein gM of pseudorabies virus results in attenuation for the natural host. J. Gen. Virol. 1997, 78 Pt 9, 2147–2151. [Google Scholar] [CrossRef] [Green Version]
- Crump, C.M.; Bruun, B.; Bell, S.; Pomeranz, L.E.; Minson, T.; Browne, H.M. Alphaherpesvirus glycoprotein M causes the relocalization of plasma membrane proteins. J. Gen. Virol. 2004, 85 Pt 12, 3517–3527. [Google Scholar] [CrossRef]
- Pannhorst, K.; Wei, H.; Yezid, H.; He, J.; Chowdhury, S.I. Bovine Herpesvirus 1 U(L)49.5 Interacts with gM and VP22 To Ensure Virus Cell-to-Cell Spread and Virion Incorporation: Novel Role for VP22 in gM-Independent U(L)49.5 Virion Incorporation. J. Virol. 2018, 92, e00240-18. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, Y.; Sadaoka, T.; Yoshii, H.; Somboonthum, P.; Imazawa, T.; Nagaike, K.; Ozono, K.; Yamanishi, K.; Mori, Y. Varicella-zoster virus glycoprotein M homolog is glycosylated, is expressed on the viral envelope, and functions in virus cell-to-cell spread. J. Virol. 2008, 82, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.; Just, F.T.; Lischewski, A.; Elbers, K.; Neubauer, A. A glycoprotein M-deleted equid herpesvirus 4 is severely impaired in virus egress and cell-to-cell spread. J. Gen. Virol. 2005, 86 Pt 1, 11–21. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | Forward Primer | Reverse |
|---|---|---|
| pHA-gM (70–438aa) | 3′ATTTCTAGAATGGGCGCGCGCCACCCGGCGCT5′ | 3′ATGATATCTTTGACGTGCGCGGGGGGTGGG5′ |
| pHA-gM (140–438aa) | 3′ATTTCTAGAATGACCGCCGGGCTGCCCGGCGC5′ | 3′ATGATATCTTTGACGTGCGCGGGGGGTGGG5′ |
| pHA-gM (210–438aa) | 3′ATTTCTAGAATGCTGGGGCTGTCGCTGGCACA5′ | 3′ATGATATCTTTGACGTGCGCGGGGGGTGGG5′ |
| pHA-gM (300–438aa) | 3′ATTTCTAGAATGGCCCCCCGGGCTGCCGCTAG5′ | 3′ATGATATCTTTGACGTGCGCGGGGGGTGGG5′ |
| pHA-gM (250–438aa) | 3′ATTTCTAGATGGTCGCCGGCGTGACGG5′ | 3′ATGATATCTTTGACGTGCGCGGGGGGTGGG5′ |
| pHA-gM (140–264aa) | 3′ATTTCTAGATGGCGCTGGCGGCCT5′ | 3′ATGATATCTTTCAAAAAGAGCACGGCGG5′ |
| Primer Name | Primer Sequence |
|---|---|
| Primer 1 | 3′GGAGGTACCGTATTACCGCCATGCATTAG5′ |
| Primer 2 | 3′GAGGGATCCTGCCGATTTCGGCCTATTGG5′ |
| Primer 3 | 3′GCGGAATTCGCGCTGCATCTCGTCACTTTCATCG5′ |
| Primer 4 | 3′GAAGGTACCCGCCAACCATACCGCTAAGGAGACC5′ |
| Primer 5 | 3′GAGAAGCTTCGTAAAGCTGCGCCGACAGGAG5′ |
| Primer 6 | 3′CTGCGCGGAGCCCGCGATGACGGCAAC5′ |
| Primer 7 | 3′GAAGGATCCCGCCAACCATACCGCTAAGGAGACC5′ |
| Primer 8 | 3′GGCGTTGCCGTCATCGCGGGCTCCGCGCAG5′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sucharita, S.; Tikoo, S.; van Drunen Littel-van den Hurk, S. Bovine Herpesvirus-1 Glycoprotein M Mediates the Translocation to the Golgi Apparatus and Packaging of VP8. Viruses 2022, 14, 1985. https://doi.org/10.3390/v14091985
Sucharita S, Tikoo S, van Drunen Littel-van den Hurk S. Bovine Herpesvirus-1 Glycoprotein M Mediates the Translocation to the Golgi Apparatus and Packaging of VP8. Viruses. 2022; 14(9):1985. https://doi.org/10.3390/v14091985
Chicago/Turabian StyleSucharita, Soumya, Suresh Tikoo, and Sylvia van Drunen Littel-van den Hurk. 2022. "Bovine Herpesvirus-1 Glycoprotein M Mediates the Translocation to the Golgi Apparatus and Packaging of VP8" Viruses 14, no. 9: 1985. https://doi.org/10.3390/v14091985