
Mechanistic Interplay between HIV-1 Reverse Transcriptase Enzyme Kinetics and Host SAMHD1 Protein: Viral Myeloid-Cell Tropism and Genomic Mutagenesis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Replication Kinetics of HIV-1 in Target Cells
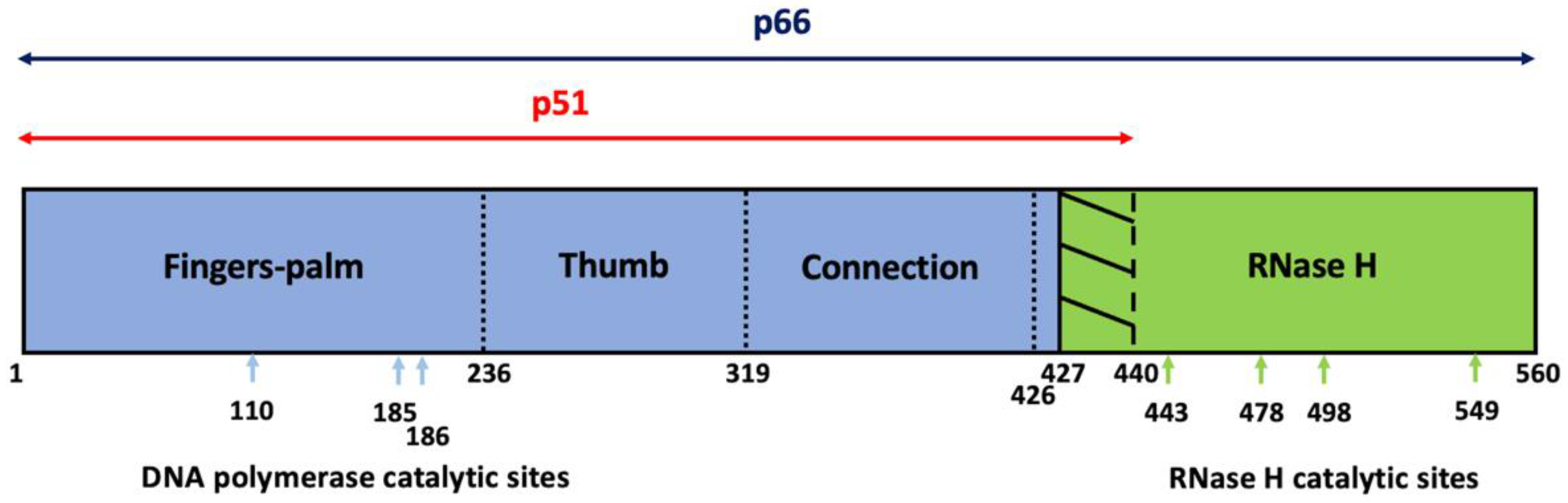
3. Structural and Enzymological Background of HIV-1 RT
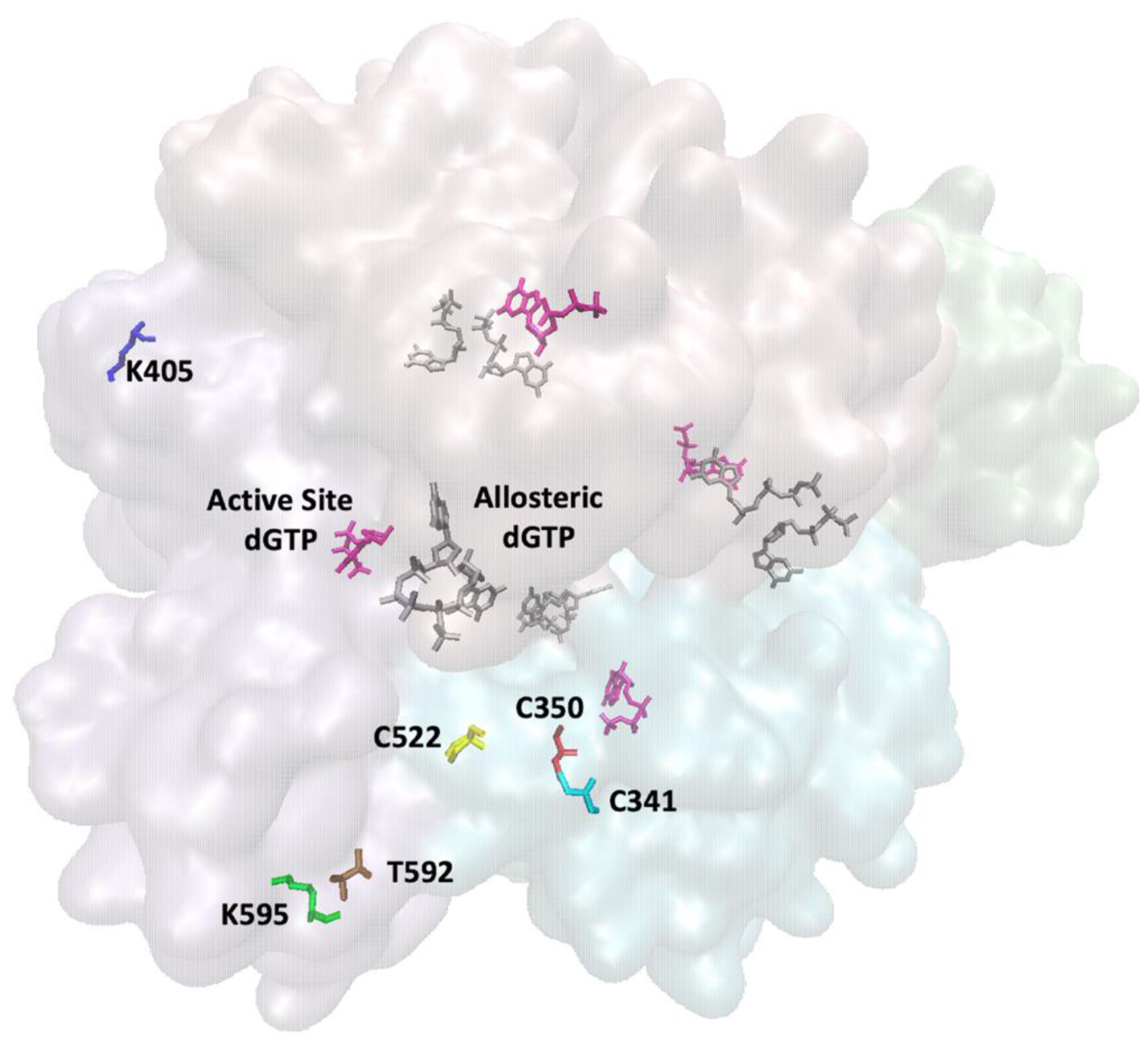
4. HIV-1 RT Kinetics
5. The Fidelity Profiles of HIV-1 RT
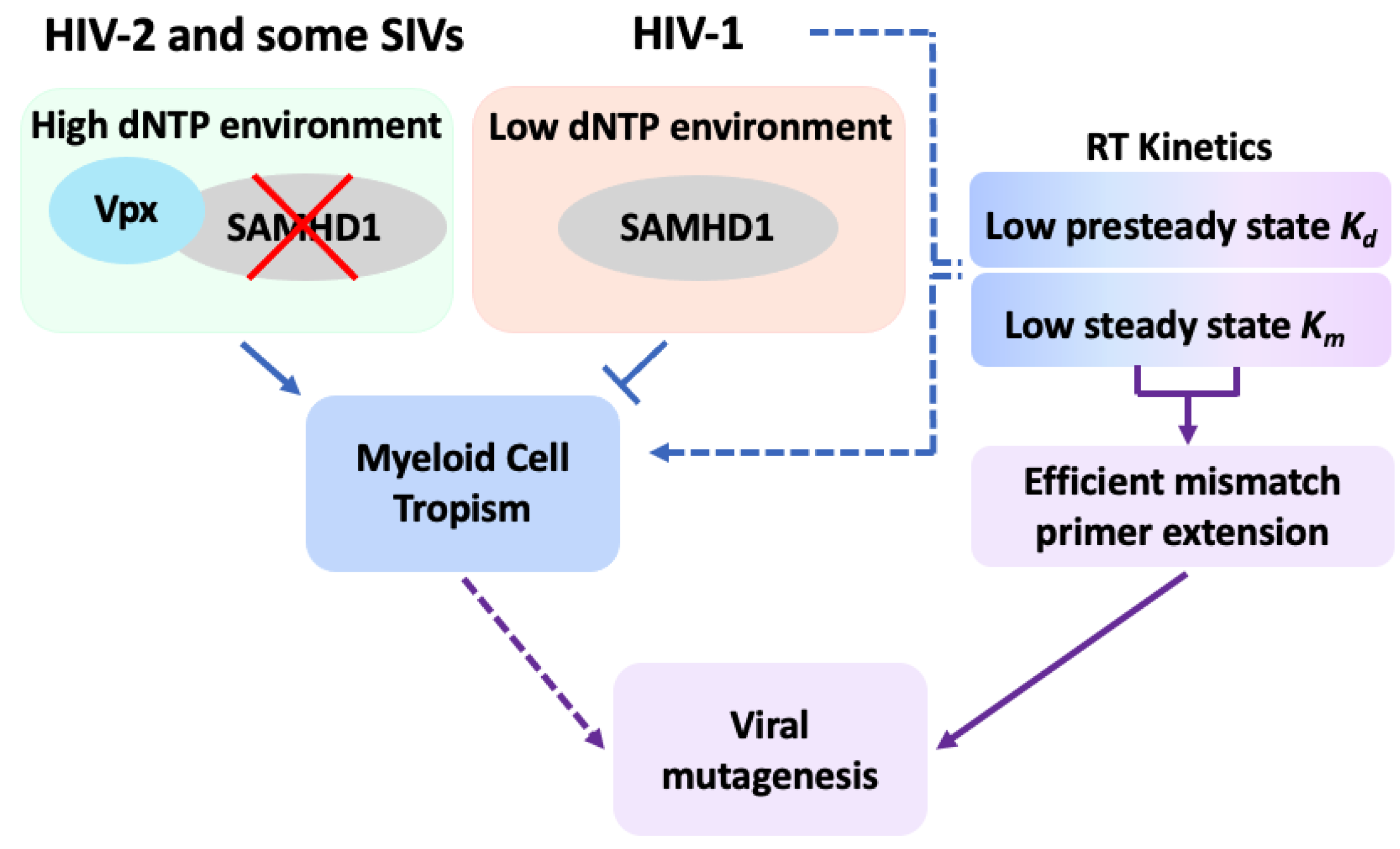
6. Regulation of Cellular dNTP Pool as a Major Determinant of HIV-1 Replication Kinetics in Viral Target Cells
7. Regulation of Human SAMHD1
8. HIV-1 Restriction by Human SAMHD1
9. Viral Protein X (Vpx) Counteracts Lentiviral Restriction by SAMHD1
10. Host and Viral Proteins’ Evolution Due to the Host–Pathogen Arms Race
11. SAMHD1 and Host Innate Immunity
12. SAMHD1: Other Considerations
13. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zheng, J.; Wei, Y.; Han, G.Z. The diversity and evolution of retroviruses: Perspectives from viral “fossils”. Virol. Sin. 2022, 37, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.H. Reverse Transcription of Retroviruses and LTR Retrotransposons. Microbiol. Spectr. 2015, 3, 1051–1077. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Primers 2015, 1, 15035. [Google Scholar] [CrossRef]
- Kennedy, E.M.; Gavegnano, C.; Nguyen, L.; Slater, R.; Lucas, A.; Fromentin, E.; Schinazi, R.F.; Kim, B. Ribonucleoside triphosphates as substrate of human immunodeficiency virus type 1 reverse transcriptase in human macrophages. J. Biol. Chem. 2010, 285, 39380–39391. [Google Scholar] [CrossRef] [Green Version]
- Diamond, T.L.; Roshal, M.; Jamburuthugoda, V.K.; Reynolds, H.M.; Merriam, A.R.; Lee, K.Y.; Balakrishnan, M.; Bambara, R.A.; Planelles, V.; Dewhurst, S.; et al. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J. Biol. Chem. 2004, 279, 51545–51553. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.D.; Bebenek, K.; Kunkel, T.A. The accuracy of reverse transcriptase from HIV-1. Science 1988, 242, 1171–1173. [Google Scholar] [CrossRef]
- Stultz, R.D.; Cenker, J.J.; McDonald, D. Imaging HIV-1 genomic DNA from entry through productive infection. J. Virol. 2017, 91, e00034-17. [Google Scholar] [CrossRef] [Green Version]
- Franzolin, E.; Pontarin, G.; Rampazzo, C.; Miazzi, C.; Ferraro, P.; Palumbo, E.; Reichard, P.; Bianchi, V. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 14272–14277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef]
- Guyader, M.; Emerman, M.; Montagnier, L.; Peden, K. VPX mutants of HIV-2 are infectious in established cell lines but display a severe defect in peripheral blood lymphocytes. EMBO J. 1989, 8, 1169–1175. [Google Scholar] [CrossRef]
- Yu, X.F.; Yu, Q.C.; Essex, M.; Lee, T.H. The vpx gene of simian immunodeficiency virus facilitates efficient viral replication in fresh lymphocytes and macrophage. J. Virol. 1991, 65, 5088–5091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Lenzi, G.M.; Domaoal, R.A.; Kim, D.H.; Schinazi, R.F.; Kim, B. Kinetic variations between reverse transcriptases of viral protein X coding and noncoding lentiviruses. Retrovirology 2014, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Collin, M.; Gordon, S. The kinetics of human immunodeficiency virus reverse transcription are slower in primary human macrophages than in a lymphoid cell line. Virology 1994, 200, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, D.A.; Puertas, M.C.; Börner, K.; Martinez-Picado, J.; Müller, B.; Kräusslich, H.G. Detailed Characterization of Early HIV-1 Replication Dynamics in Primary Human Macrophages. Viruses 2018, 10, 620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruize, Z.; Kootstra, N.A. The Role of Macrophages in HIV-1 Persistence and Pathogenesis. Front. Microbiol. 2019, 10, 2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veenhuis, R.T.; Abreu, C.M.; Shirk, E.N.; Gama, L.; Clements, J.E. HIV replication and latency in monocytes and macrophages. Semin. Immunol. 2021, 51, 101472. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.E.; Jaworowski, A.; Hearps, A.C. The HIV Reservoir in Monocytes and Macrophages. Front. Immunol. 2019, 10, 1435. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, T.; Brown, C.R.; Endo, Y.; Buckler-White, A.; Plishka, R.; Bischofberger, N.; Hirsch, V.; Martin, M.A. Macrophage are the principal reservoir and sustain high virus loads in rhesus macaques after the depletion of CD4+ T cells by a highly pathogenic simian immunodeficiency virus/HIV type 1 chimera (SHIV): Implications for HIV-1 infections of humans. Proc. Natl. Acad. Sci. USA 2001, 98, 658–663. [Google Scholar] [CrossRef]
- Sharaf, N.G.; Poliner, E.; Slack, R.L.; Christen, M.T.; Byeon, I.J.; Parniak, M.A.; Gronenborn, A.M.; Ishima, R. The p66 immature precursor of HIV-1 reverse transcriptase. Proteins 2014, 82, 2343–2352. [Google Scholar] [CrossRef] [Green Version]
- Sluis-Cremer, N.; Arion, D.; Abram, M.E.; Parniak, M.A. Proteolytic processing of an HIV-1 pol polyprotein precursor: Insights into the mechanism of reverse transcriptase p66/p51 heterodimer formation. Int. J. Biochem. Cell Biol. 2004, 36, 1836–1847. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L.; Betancor, G.; Matamoros, T. HIV-1 reverse transcriptase connection subdomain mutations involved in resistance to approved non-nucleoside inhibitors. Antivir. Res. 2011, 92, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Tomasselli, A.G.; Sarcich, J.L.; Barrett, L.J.; Reardon, I.M.; Howe, W.J.; Evans, D.B.; Sharma, S.K.; Heinrikson, R.L. Human immunodeficiency virus type-1 reverse transcriptase and ribonuclease H as substrates of the viral protease. Protein Sci. 1993, 2, 2167–2176. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Smerdon, S.J.; Jäger, J.; Kohlstaedt, L.A.; Rice, P.A.; Friedman, J.M.; Steitz, T.A. Structural basis of asymmetry in the human immunodeficiency virus type 1 reverse transcriptase heterodimer. Proc. Natl. Acad. Sci. USA 1994, 91, 7242–7246. [Google Scholar] [CrossRef] [Green Version]
- Jacobo-Molina, A.; Ding, J.; Nanni, R.G.; Clark, A.D., Jr.; Lu, X.; Tantillo, C.; Williams, R.L.; Kamer, G.; Ferris, A.L.; Clark, P.; et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc. Natl. Acad. Sci. USA 1993, 90, 6320–6324. [Google Scholar] [CrossRef] [Green Version]
- Larder, B.A.; Purifoy, D.J.; Powell, K.L.; Darby, G. Site-specific mutagenesis of AIDS virus reverse transcriptase. Nature 1987, 327, 716–717. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.M.; Parmar, V.; Kemp, S.D.; Larder, B.A. Mutational analysis of two conserved sequence motifs in HIV-1 reverse transcriptase. FEBS Lett. 1991, 282, 231–234. [Google Scholar] [CrossRef] [Green Version]
- Mizrahi, V.; Brooksbank, R.L.; Nkabinde, N.C. Mutagenesis of the conserved aspartic acid 443, glutamic acid 478, asparagine 494, and aspartic acid 498 residues in the ribonuclease H domain of p66/p51 human immunodeficiency virus type I reverse transcriptase. Expression and biochemical analysis. J. Biol. Chem. 1994, 269, 19245–19249. [Google Scholar] [CrossRef]
- Álvarez, M.; Barrioluengo, V.; Afonso-Lehmann, R.N.; Menéndez-Arias, L. Altered error specificity of RNase H-deficient HIV-1 reverse transcriptases during DNA-dependent DNA synthesis. Nucleic Acids Res. 2013, 41, 4601–4612. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science 1998, 282, 1669–1675. [Google Scholar] [CrossRef] [Green Version]
- Ha, B.; Larsen, K.P.; Zhang, J.; Fu, Z.; Montabana, E.; Jackson, L.N.; Chen, D.H.; Puglisi, E.V. High-resolution view of HIV-1 reverse transcriptase initiation complexes and inhibition by NNRTI drugs. Nat. Commun. 2021, 12, 2500. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.P.; Mathiharan, Y.K.; Kappel, K.; Coey, A.T.; Chen, D.H.; Barrero, D.; Madigan, L.; Puglisi, J.D.; Skiniotis, G.; Puglisi, E.V. Architecture of an HIV-1 reverse transcriptase initiation complex. Nature 2018, 557, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Beerens, N.; Berkhout, B. The tRNA primer activation signal in the human immunodeficiency virus type 1 genome is important for initiation and processive elongation of reverse transcription. J. Virol. 2002, 76, 2329–2339. [Google Scholar] [CrossRef] [Green Version]
- Beerens, N.; Groot, F.; Berkhout, B. Initiation of HIV-1 reverse transcription is regulated by a primer activation signal. J. Biol. Chem. 2001, 276, 31247–31256. [Google Scholar] [CrossRef] [Green Version]
- Goldschmidt, V.; Ehresmann, C.; Ehresmann, B.; Marquet, R. Does the HIV-1 primer activation signal interact with tRNA3(Lys) during the initiation of reverse transcription? Nucleic Acids Res. 2003, 31, 850–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldschmidt, V.; Rigourd, M.; Ehresmann, C.; Le Grice, S.F.; Ehresmann, B.; Marquet, R. Direct and indirect contributions of RNA secondary structure elements to the initiation of HIV-1 reverse transcription. J. Biol. Chem. 2002, 277, 43233–43242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isel, C.; Westhof, E.; Massire, C.; Le Grice, S.F.; Ehresmann, B.; Ehresmann, C.; Marquet, R. Structural basis for the specificity of the initiation of HIV-1 reverse transcription. EMBO J. 1999, 18, 1038–1048. [Google Scholar] [CrossRef] [Green Version]
- Isel, C.; Lanchy, J.M.; Le Grice, S.F.; Ehresmann, C.; Ehresmann, B.; Marquet, R. Specific initiation and switch to elongation of human immunodeficiency virus type 1 reverse transcription require the post-transcriptional modifications of primer tRNA3Lys. EMBO J. 1996, 15, 917–924. [Google Scholar] [CrossRef]
- Lanchy, J.M.; Ehresmann, C.; Le Grice, S.F.; Ehresmann, B.; Marquet, R. Binding and kinetic properties of HIV-1 reverse transcriptase markedly differ during initiation and elongation of reverse transcription. EMBO J. 1996, 15, 7178–7187. [Google Scholar] [CrossRef]
- Liu, S.; Harada, B.T.; Miller, J.T.; Le Grice, S.F.; Zhuang, X. Initiation complex dynamics direct the transitions between distinct phases of early HIV reverse transcription. Nat. Struct. Mol. Biol. 2010, 17, 1453–1460. [Google Scholar] [CrossRef] [Green Version]
- Skasko, M.; Weiss, K.K.; Reynolds, H.M.; Jamburuthugoda, V.; Lee, K.; Kim, B. Mechanistic differences in RNA-dependent DNA polymerization and fidelity between murine leukemia virus and HIV-1 reverse transcriptases. J. Biol. Chem. 2005, 280, 12190–12200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kati, W.M.; Johnson, K.A.; Jerva, L.F.; Anderson, K.S. Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 1992, 267, 25988–25997. [Google Scholar] [CrossRef]
- DeStefano, J.J.; Buiser, R.G.; Mallaber, L.M.; Myers, T.W.; Bambara, R.A.; Fay, P.J. Polymerization and RNase H activities of the reverse transcriptases from avian myeloblastosis, human immunodeficiency, and Moloney murine leukemia viruses are functionally uncoupled. J. Biol. Chem. 1991, 266, 7423–7431. [Google Scholar] [CrossRef]
- Figiel, M.; Krepl, M.; Poznanski, J.; Golab, A.; Šponer, J.; Nowotny, M. Coordination between the polymerase and RNase H activity of HIV-1 reverse transcriptase. Nucleic Acids Res. 2017, 45, 3341–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achuthan, V.; Keith, B.J.; Connolly, B.A.; DeStefano, J.J. Human immunodeficiency virus reverse transcriptase displays dramatically higher fidelity under physiological magnesium conditions in vitro. J. Virol. 2014, 88, 8514–8527. [Google Scholar] [CrossRef] [Green Version]
- Preston, B.D.; Poiesz, B.J.; Loeb, L.A. Fidelity of HIV-1 reverse transcriptase. Science 1988, 242, 1168–1171. [Google Scholar] [CrossRef]
- Rezende, L.F.; Kew, Y.; Prasad, V.R. The effect of increased processivity on overall fidelity of human immunodeficiency virus type 1 reverse transcriptase. J. Biomed. Sci. 2001, 8, 197–205. [Google Scholar] [CrossRef]
- Kaushik, N.; Rege, N.; Yadav, P.N.; Sarafianos, S.G.; Modak, M.J.; Pandey, V.N. Biochemical analysis of catalytically crucial aspartate mutants of human immunodeficiency virus type 1 reverse transcriptase. Biochemistry 1996, 35, 11536–11546. [Google Scholar] [CrossRef]
- Goldschmidt, V.; Didierjean, J.; Ehresmann, B.; Ehresmann, C.; Isel, C.; Marquet, R. Mg2+ dependency of HIV-1 reverse transcription, inhibition by nucleoside analogues and resistance. Nucleic Acids Res. 2006, 34, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Dilmore, C.R.; DeStefano, J.J. HIV Reverse Transcriptase Pre-Steady-State Kinetic Analysis of Chain Terminators and Translocation Inhibitors Reveals Interactions between Magnesium and Nucleotide 3’-OH. ACS Omega 2021, 6, 14621–14628. [Google Scholar] [CrossRef]
- Yokoyama, M.; Mori, H.; Sato, H. Allosteric regulation of HIV-1 reverse transcriptase by ATP for nucleotide selection. PLoS ONE 2010, 5, e8867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawson, J.M.O.; Gohl, D.M.; Landman, S.R.; Roth, M.E.; Meissner, M.E.; Peterson, T.S.; Hodges, J.S.; Beckman, K.B.; Mansky, L.M. Single-Strand Consensus Sequencing Reveals that HIV Type but not Subtype Significantly Impacts Viral Mutation Frequencies and Spectra. J. Mol. Biol. 2017, 429, 2290–2307. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, J.M.; Geller, R.; Garijo, R.; López-Aldeguer, J.; Sanjuán, R. Extremely High Mutation Rate of HIV-1 In Vivo. PLoS Biol. 2015, 13, e1002251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Mudry, R.A., Jr.; Rexrode, C.A., 2nd; Pathak, V.K. Retroviral mutation rates and A-to-G hypermutations during different stages of retroviral replication. J. Virol. 1996, 70, 7594–7602. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.D.; Preston, B.D.; Johnston, L.A.; Soni, A.; Loeb, L.A.; Kunkel, T.A. Fidelity of two retroviral reverse transcriptases during DNA-dependent DNA synthesis in vitro. Mol. Cell Biol. 1989, 9, 469–476. [Google Scholar]
- Menéndez-Arias, L. Mutation rates and intrinsic fidelity of retroviral reverse transcriptases. Viruses 2009, 1, 1137–1165. [Google Scholar] [CrossRef] [Green Version]
- Martín-Alonso, S.; Frutos-Beltrán, E.; Menéndez-Arias, L. Reverse Transcriptase: From Transcriptomics to Genome Editing. Trends Biotechnol. 2021, 39, 194–210. [Google Scholar] [CrossRef]
- Garforth, S.J.; Domaoal, R.A.; Lwatula, C.; Landau, M.J.; Meyer, A.J.; Anderson, K.S.; Prasad, V.R. K65R and K65A substitutions in HIV-1 reverse transcriptase enhance polymerase fidelity by decreasing both dNTP misinsertion and mispaired primer extension efficiencies. J. Mol. Biol. 2010, 401, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Lwatula, C.; Garforth, S.J.; Prasad, V.R. Lys66 residue as a determinant of high mismatch extension and misinsertion rates of HIV-1 reverse transcriptase. FEBS J. 2012, 279, 4010–4024. [Google Scholar] [CrossRef] [Green Version]
- Shah, F.S.; Curr, K.A.; Hamburgh, M.E.; Parniak, M.; Mitsuya, H.; Arnez, J.G.; Prasad, V.R. Differential influence of nucleoside analog-resistance mutations K65R and L74V on the overall mutation rate and error specificity of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 2000, 275, 27037–27044. [Google Scholar] [CrossRef]
- Barrioluengo, V.; Alvarez, M.; Barbieri, D.; Menéndez-Arias, L. Thermostable HIV-1 group O reverse transcriptase variants with the same fidelity as murine leukaemia virus reverse transcriptase. Biochem. J. 2011, 436, 599–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez, M.; Sebastián-Martín, A.; García-Marquina, G.; Menéndez-Arias, L. Fidelity of classwide-resistant HIV-2 reverse transcriptase and differential contribution of K65R to the accuracy of HIV-1 and HIV-2 reverse transcriptases. Sci. Rep. 2017, 7, 44834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, J.C.; Bebenek, K.; Kunkel, T.A. Unequal human immunodeficiency virus type 1 reverse transcriptase error rates with RNA and DNA templates. Proc. Natl. Acad. Sci. USA 1992, 89, 6919–6923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.P.; Loeb, L.A. Fidelity of HIV-1 reverse transcriptase copying RNA in vitro. Biochemistry 1992, 31, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Loeb, L.A. Fidelity of HIV-1 reverse transcriptase copying a hypervariable region of the HIV-1 env gene. Virology 1994, 199, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.E.; Ferris, A.L.; Shao, W.; Alvord, W.G.; Hughes, S.H. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 2010, 84, 9864–9878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelman, L.V.; Petruska, J.; Goodman, M.F. Base mispair extension kinetics. Comparison of DNA polymerase alpha and reverse transcriptase. J. Biol. Chem. 1990, 265, 2338–2346. [Google Scholar] [CrossRef]
- Geller, R.; Domingo-Calap, P.; Cuevas, J.M.; Rossolillo, P.; Negroni, M.; Sanjuán, R. The external domains of the HIV-1 envelope are a mutational cold spot. Nat. Commun. 2015, 6, 8571. [Google Scholar] [CrossRef] [Green Version]
- Yeo, J.Y.; Koh, D.W.; Yap, P.; Goh, G.R.; Gan, S.K. Spontaneous Mutations in HIV-1 Gag, Protease, RT p66 in the First Replication Cycle and How They Appear: Insights from an In Vitro Assay on Mutation Rates and Types. Int. J. Mol. Sci. 2020, 22, 370. [Google Scholar] [CrossRef]
- Greene, B.L.; Kang, G.; Cui, C.; Bennati, M.; Nocera, D.G.; Drennan, C.L.; Stubbe, J. Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets. Annu. Rev. Biochem. 2020, 89, 45–75. [Google Scholar] [CrossRef]
- Young, J.D.; Yao, S.Y.; Baldwin, J.M.; Cass, C.E.; Baldwin, S.A. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Aspects Med. 2013, 34, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Kashlan, O.B.; Scott, C.P.; Lear, J.D.; Cooperman, B.S. A comprehensive model for the allosteric regulation of mammalian ribonucleotide reductase. Functional consequences of ATP- and dATP-induced oligomerization of the large subunit. Biochemistry 2002, 41, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Engström, Y.; Eriksson, S.; Jildevik, I.; Skog, S.; Thelander, L.; Tribukait, B. Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J. Biol. Chem. 1985, 260, 9114–9116. [Google Scholar] [CrossRef]
- Eriksson, S.; Gräslund, A.; Skog, S.; Thelander, L.; Tribukait, B. Cell cycle-dependent regulation of mammalian ribonucleotide reductase. The S phase-correlated increase in subunit M2 is regulated by de novo protein synthesis. J. Biol. Chem. 1984, 259, 11695–11700. [Google Scholar] [CrossRef]
- Björklund, S.; Skog, S.; Tribukait, B.; Thelander, L. S-phase-specific expression of mammalian ribonucleotide reductase R1 and R2 subunit mRNAs. Biochemistry 1990, 29, 5452–5458. [Google Scholar] [CrossRef]
- Yao, R.; Zhang, Z.; An, X.; Bucci, B.; Perlstein, D.L.; Stubbe, J.; Huang, M. Subcellular localization of yeast ribonucleotide reductase regulated by the DNA replication and damage checkpoint pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 6628–6633. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Zhou, Z.; Elledge, S.J. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 1998, 94, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Coppock, D.L.; Pardee, A.B. Control of thymidine kinase mRNA during the cell cycle. Mol. Cell Biol. 1987, 7, 2925–2932. [Google Scholar]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Nguyen, L.A.; Daddacha, W.; Hollenbaugh, J.A. Tight interplay among SAMHD1 protein level, cellular dNTP levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J. Biol. Chem. 2012, 287, 21570–21574. [Google Scholar] [CrossRef] [Green Version]
- Brandariz-Nuñez, A.; Valle-Casuso, J.C.; White, T.E.; Laguette, N.; Benkirane, M.; Brojatsch, J.; Diaz-Griffero, F. Role of SAMHD1 nuclear localization in restriction of HIV-1 and SIVmac. Retrovirology 2012, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaller, T.; Pollpeter, D.; Apolonia, L.; Goujon, C.; Malim, M.H. Nuclear import of SAMHD1 is mediated by a classical karyopherin α/β1 dependent pathway and confers sensitivity to VpxMAC induced ubiquitination and proteasomal degradation. Retrovirology 2014, 11, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, H.; Logue, E.C.; Bloch, N.; Daddacha, W.; Polsky, S.B.; Schultz, M.L.; Kim, B.; Landau, N.R. The Vpx lentiviral accessory protein targets SAMHD1 for degradation in the nucleus. J. Virol. 2012, 86, 12552–12560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.E.; Brandariz-Nunez, A.; Valle-Casuso, J.C.; Amie, S.; Nguyen, L.; Kim, B.; Brojatsch, J.; Diaz-Griffero, F. Contribution of SAM and HD domains to retroviral restriction mediated by human SAMHD1. Virology 2013, 436, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Aravind, L.; Koonin, E.V. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 1998, 23, 469–472. [Google Scholar] [CrossRef]
- Cribier, A.; Descours, B.; Valadao, A.L.; Laguette, N.; Benkirane, M. Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell Rep. 2013, 3, 1036–1043. [Google Scholar] [CrossRef] [Green Version]
- St Gelais, C.; Kim, S.H.; Ding, L.; Yount, J.S.; Ivanov, D.; Spearman, P.; Wu, L. A Putative Cyclin-binding Motif in Human SAMHD1 Contributes to Protein Phosphorylation, Localization, and Stability. J. Biol. Chem. 2016, 291, 26332–26342. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Hao, C.; DeLucia, M.; Swanson, S.; Florens, L.; Washburn, M.P.; Ahn, J.; Skowronski, J. CyclinA2-Cyclin-dependent Kinase Regulates SAMHD1 Protein Phosphohydrolase Domain. J. Biol. Chem. 2015, 290, 13279–13292. [Google Scholar] [CrossRef] [Green Version]
- Schott, K.; Fuchs, N.V.; Derua, R.; Mahboubi, B.; Schnellbächer, E.; Seifried, J.; Tondera, C.; Schmitz, H.; Shepard, C.; Brandariz-Nuñez, A.; et al. Dephosphorylation of the HIV-1 restriction factor SAMHD1 is mediated by PP2A-B55α holoenzymes during mitotic exit. Nat. Commun. 2018, 9, 2227. [Google Scholar] [CrossRef]
- Zhu, C.; Gao, W.; Zhao, K.; Qin, X.; Zhang, Y.; Peng, X.; Zhang, L.; Dong, Y.; Zhang, W.; Li, P.; et al. Structural insight into dGTP-dependent activation of tetrameric SAMHD1 deoxynucleoside triphosphate triphosphohydrolase. Nat. Commun. 2013, 4, 2722. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Kaur, S.; DeLucia, M.; Hao, C.; Mehrens, J.; Wang, C.; Golczak, M.; Palczewski, K.; Gronenborn, A.M.; Ahn, J.; et al. Tetramerization of SAMHD1 is required for biological activity and inhibition of HIV infection. J. Biol. Chem. 2013, 288, 10406–10417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, X.; Tang, C.; Zhao, Q.; Wang, W.; Xiong, Y. Structural basis of cellular dNTP regulation by SAMHD1. Proc. Natl. Acad. Sci. USA 2014, 111, E4305–E4314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, X.; Wu, Y.; Yan, J.; Mehrens, J.; Yang, H.; DeLucia, M.; Hao, C.; Gronenborn, A.M.; Skowronski, J.; Ahn, J.; et al. Mechanism of allosteric activation of SAMHD1 by dGTP. Nat. Struct. Mol. Biol. 2013, 20, 1304–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, R.D.; Holland, P.J.; Hollis, T.; Perrino, F.W. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J. Biol. Chem. 2011, 286, 43596–43600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amie, S.M.; Bambara, R.A.; Kim, B. GTP is the primary activator of the anti-HIV restriction factor SAMHD1. J. Biol. Chem. 2013, 288, 25001–25006. [Google Scholar] [CrossRef] [Green Version]
- Koharudin, L.M.; Wu, Y.; DeLucia, M.; Mehrens, J.; Gronenborn, A.M.; Ahn, J. Structural basis of allosteric activation of sterile α motif and histidine-aspartate domain-containing protein 1 (SAMHD1) by nucleoside triphosphates. J. Biol. Chem. 2014, 289, 32617–32627. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.K.; Bhattacharya, A.; Bhattacharya, S. Uncovering allostery and regulation in SAMHD1 through molecular dynamics simulations. Proteins 2017, 85, 1266–1275. [Google Scholar] [CrossRef]
- Hansen, E.C.; Seamon, K.J.; Cravens, S.L.; Stivers, J.T. GTP activator and dNTP substrates of HIV-1 restriction factor SAMHD1 generate a long-lived activated state. Proc. Natl. Acad. Sci. USA 2014, 111, E1843–E1851. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Bhattacharya, A.; Villacorta, J.; Diaz-Griffero, F.; Ivanov, D.N. Allosteric Activation of SAMHD1 Protein by Deoxynucleotide Triphosphate (dNTP)-dependent Tetramerization Requires dNTP Concentrations That Are Similar to dNTP Concentrations Observed in Cycling T Cells. J. Biol. Chem. 2016, 291, 21407–21413. [Google Scholar] [CrossRef] [Green Version]
- Arnold, L.H.; Groom, H.C.; Kunzelmann, S.; Schwefel, D.; Caswell, S.J.; Ordonez, P.; Mann, M.C.; Rueschenbaum, S.; Goldstone, D.C.; Pennell, S.; et al. Phospho-dependent Regulation of SAMHD1 Oligomerisation Couples Catalysis and Restriction. PLoS Pathog. 2015, 11, e1005194. [Google Scholar] [CrossRef]
- Hollenbaugh, J.A.; Shelton, J.; Tao, S.; Amiralaei, S.; Liu, P.; Lu, X.; Goetze, R.W.; Zhou, L.; Nettles, J.H.; Schinazi, R.F.; et al. Substrates and Inhibitors of SAMHD1. PLoS ONE 2017, 12, e0169052. [Google Scholar] [CrossRef] [PubMed]
- Herold, N.; Rudd, S.G.; Ljungblad, L.; Sanjiv, K.; Myrberg, I.H.; Paulin, C.B.; Heshmati, Y.; Hagenkort, A.; Kutzner, J.; Page, B.D.; et al. Targeting SAMHD1 with the Vpx protein to improve cytarabine therapy for hematological malignancies. Nat. Med. 2017, 23, 256–263. [Google Scholar] [CrossRef]
- Schneider, C.; Oellerich, T.; Baldauf, H.M.; Schwarz, S.M.; Thomas, D.; Flick, R.; Bohnenberger, H.; Kaderali, L.; Stegmann, L.; Cremer, A.; et al. SAMHD1 is a biomarker for cytarabine response and a therapeutic target in acute myeloid leukemia. Nat. Med. 2017, 23, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Amie, S.M.; Daly, M.B.; Noble, E.; Schinazi, R.F.; Bambara, R.A.; Kim, B. Anti-HIV host factor SAMHD1 regulates viral sensitivity to nucleoside reverse transcriptase inhibitors via modulation of cellular deoxyribonucleoside triphosphate (dNTP) levels. J. Biol. Chem. 2013, 288, 20683–20691. [Google Scholar] [CrossRef] [Green Version]
- Huber, A.D.; Michailidis, E.; Schultz, M.L.; Ong, Y.T.; Bloch, N.; Puray-Chavez, M.N.; Leslie, M.D.; Ji, J.; Lucas, A.D.; Kirby, K.A.; et al. SAMHD1 has differential impact on the efficacies of HIV nucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2014, 58, 4915–4919. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.R.; Caswell, S.J.; Kunzelmann, S.; Arnold, L.H.; Purkiss, A.G.; Kelly, G.; Taylor, I.A. Crystal structures of SAMHD1 inhibitor complexes reveal the mechanism of water-mediated dNTP hydrolysis. Nat. Commun. 2020, 11, 3165. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Mansour, M.N.; Dillman, K.S.; Perez, J.R.; Danley, D.E.; Aeed, P.A.; Simons, S.P.; Lemotte, P.K.; Menniti, F.S. Structural basis for the catalytic mechanism of human phosphodiesterase 9. Proc. Natl. Acad. Sci. USA 2008, 105, 13309–13314. [Google Scholar] [CrossRef] [Green Version]
- Rinaldo, S.; Paiardini, A.; Stelitano, V.; Brunotti, P.; Cervoni, L.; Fernicola, S.; Protano, C.; Vitali, M.; Cutruzzolà, F.; Giardina, G. Structural basis of functional diversification of the HD-GYP domain revealed by the Pseudomonas aeruginosa PA4781 protein, which displays an unselective bimetallic binding site. J. Bacteriol. 2015, 197, 1525–1535. [Google Scholar] [CrossRef] [Green Version]
- Huynh, T.N.; Luo, S.; Pensinger, D.; Sauer, J.D.; Tong, L.; Woodward, J.J. An HD-domain phosphodiesterase mediates cooperative hydrolysis of c-di-AMP to affect bacterial growth and virulence. Proc. Natl. Acad. Sci. USA 2015, 112, E747–E756. [Google Scholar] [CrossRef] [Green Version]
- White, T.E.; Brandariz-Nuñez, A.; Valle-Casuso, J.C.; Amie, S.; Nguyen, L.A.; Kim, B.; Tuzova, M.; Diaz-Griffero, F. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe 2013, 13, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Pauls, E.; Ruiz, A.; Badia, R.; Permanyer, M.; Gubern, A.; Riveira-Muñoz, E.; Torres-Torronteras, J.; Alvarez, M.; Mothe, B.; Brander, C.; et al. Cell cycle control and HIV-1 susceptibility are linked by CDK6-dependent CDK2 phosphorylation of SAMHD1 in myeloid and lymphoid cells. J. Immunol. 2014, 193, 1988–1997. [Google Scholar] [CrossRef] [PubMed]
- Coiras, M.; Bermejo, M.; Descours, B.; Mateos, E.; García-Pérez, J.; López-Huertas, M.R.; Lederman, M.M.; Benkirane, M.; Alcamí, J. IL-7 Induces SAMHD1 Phosphorylation in CD4+ T Lymphocytes, Improving Early Steps of HIV-1 Life Cycle. Cell Rep. 2016, 14, 2100–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlcochova, P.; Sutherland, K.A.; Watters, S.A.; Bertoli, C.; de Bruin, R.A.; Rehwinkel, J.; Neil, S.J.; Lenzi, G.M.; Kim, B.; Khwaja, A.; et al. A G1-like state allows HIV-1 to bypass SAMHD1 restriction in macrophages. EMBO J. 2017, 36, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.K.; Bhattacharya, A.; Bhattacharya, S. Allosteric Signal Transduction in HIV-1 Restriction Factor SAMHD1 Proceeds via Reciprocal Handshake across Monomers. J. Chem. Inf. Model. 2017, 57, 2523–2538. [Google Scholar] [CrossRef]
- Tang, C.; Ji, X.; Wu, L.; Xiong, Y. Impaired dNTPase activity of SAMHD1 by phosphomimetic mutation of Thr-592. J. Biol. Chem. 2015, 290, 26352–26359. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, A.; Pauls, E.; Badia, R.; Torres-Torronteras, J.; Riveira-Muñoz, E.; Clotet, B.; Martí, R.; Ballana, E.; Esté, J.A. Cyclin D3-dependent control of the dNTP pool and HIV-1 replication in human macrophages. Cell Cycle 2015, 14, 1657–1665. [Google Scholar] [CrossRef]
- Pauls, E.; Badia, R.; Torres-Torronteras, J.; Ruiz, A.; Permanyer, M.; Riveira-Muñoz, E.; Clotet, B.; Marti, R.; Ballana, E.; Esté, J.A. Palbociclib, a selective inhibitor of cyclin-dependent kinase4/6, blocks HIV-1 reverse transcription through the control of sterile α motif and HD domain-containing protein-1 (SAMHD1) activity. Aids 2014, 28, 2213–2222. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Wang, Z.; White, T.; Buffone, C.; Nguyen, L.A.; Shepard, C.N.; Kim, B.; Demeler, B.; Diaz-Griffero, F.; Ivanov, D.N. Effects of T592 phosphomimetic mutations on tetramer stability and dNTPase activity of SAMHD1 can not explain the retroviral restriction defect. Sci. Rep. 2016, 6, 31353. [Google Scholar] [CrossRef] [Green Version]
- Welbourn, S.; Dutta, S.M.; Semmes, O.J.; Strebel, K. Restriction of virus infection but not catalytic dNTPase activity is regulated by phosphorylation of SAMHD1. J. Virol. 2013, 87, 11516–11524. [Google Scholar] [CrossRef] [Green Version]
- Welbourn, S.; Strebel, K. Low dNTP levels are necessary but may not be sufficient for lentiviral restriction by SAMHD1. Virology 2016, 488, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Tramentozzi, E.; Ferraro, P.; Hossain, M.; Stillman, B.; Bianchi, V.; Pontarin, G. The dNTP triphosphohydrolase activity of SAMHD1 persists during S-phase when the enzyme is phosphorylated at T592. Cell Cycle 2018, 17, 1102–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.E.; Brandariz-Nunez, A.; Martinez-Lopez, A.; Knowlton, C.; Lenzi, G.; Kim, B.; Ivanov, D.; Diaz-Griffero, F. A SAMHD1 mutation associated with Aicardi-Goutieres syndrome uncouples the ability of SAMHD1 to restrict HIV-1 from its ability to downmodulate type I interferon in humans. Hum. Mutat. 2017, 38, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Seo, J.H.; Park, J.H.; Vo, T.T.L.; An, S.; Bae, S.J.; Le, H.; Lee, H.S.; Wee, H.J.; Lee, D.; et al. SAMHD1 acetylation enhances its deoxynucleotide triphosphohydrolase activity and promotes cancer cell proliferation. Oncotarget 2017, 8, 68517–68529. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, S.; Daly, M.B.; St Gelais, C.; Kim, S.H.; Hollenbaugh, J.A.; Shepard, C.; Kennedy, E.M.; Kim, D.H.; Schinazi, R.F.; Kim, B.; et al. SAMHD1 controls cell cycle status, apoptosis and HIV-1 infection in monocytic THP-1 cells. Virology 2016, 495, 92–100. [Google Scholar] [CrossRef]
- Martinat, C.; Cormier, A.; Tobaly-Tapiero, J.; Palmic, N.; Casartelli, N.; Mahboubi, B.; Coggins, S.A.; Buchrieser, J.; Persaud, M.; Diaz-Griffero, F.; et al. SUMOylation of SAMHD1 at Lysine 595 is required for HIV-1 restriction in non-cycling cells. Nat. Commun. 2021, 12, 4582. [Google Scholar] [CrossRef]
- Kerscher, O. SUMO junction-what’s your function? New insights through SUMO-interacting motifs. EMBO Rep. 2007, 8, 550–555. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.F.; Wei, W.; Peng, X.; Dong, Y.H.; Gong, Y.; Yu, X.F. The mechanism of substrate-controlled allosteric regulation of SAMHD1 activated by GTP. Acta Crystallogr. D Biol. Crystallogr. 2015, 71 Pt 3, 516–524. [Google Scholar] [CrossRef]
- Mauney, C.H.; Rogers, L.C.; Harris, R.S.; Daniel, L.W.; Devarie-Baez, N.O.; Wu, H.; Furdui, C.M.; Poole, L.B.; Perrino, F.W.; Hollis, T. The SAMHD1 dNTP Triphosphohydrolase Is Controlled by a Redox Switch. Antioxid. Redox Signal. 2017, 27, 1317–1331. [Google Scholar] [CrossRef]
- Wang, Z.; Bhattacharya, A.; White, T.; Buffone, C.; McCabe, A.; Nguyen, L.A.; Shepard, C.N.; Pardo, S.; Kim, B.; Weintraub, S.T.; et al. Functionality of Redox-Active Cysteines Is Required for Restriction of Retroviral Replication by SAMHD1. Cell Rep. 2018, 24, 815–823. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.K.; Bhattacharya, A.; Bhattacharya, S. Molecular dynamics investigation of a redox switch in the anti-HIV protein SAMHD1. Proteins 2019, 87, 748–759. [Google Scholar] [CrossRef]
- Thapa, G.; Bhattacharya, A.; Bhattacharya, S. Dimeric Hold States of Anti-HIV Protein SAMHD1 are Redox Tunable. J. Chem. Inf. Model. 2020, 60, 6377–6391. [Google Scholar] [CrossRef]
- Batalis, S.; Rogers, L.C.; Hemphill, W.O.; Mauney, C.H.; Ornelles, D.A.; Hollis, T. SAMHD1 Phosphorylation at T592 Regulates Cellular Localization and S-phase Progression. Front. Mol. Biosci. 2021, 8, 724870. [Google Scholar] [CrossRef]
- Operario, D.J.; Balakrishnan, M.; Bambara, R.A.; Kim, B. Reduced dNTP interaction of human immunodeficiency virus type 1 reverse transcriptase promotes strand transfer. J. Biol. Chem. 2006, 281, 32113–32121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skasko, M.; Kim, B. Compensatory role of human immunodeficiency virus central polypurine tract sequence in kinetically disrupted reverse transcription. J. Virol. 2008, 82, 7716–7720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cor-Hosmer, S.K.; Daddacha, W.; Kim, B. Mechanistic interplay among the M184I HIV-1 reverse transcriptase mutant, the central polypurine tract, cellular dNTP concentrations and drug sensitivity. Virology 2010, 406, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, E.M.; Amie, S.M.; Bambara, R.A.; Kim, B. Frequent incorporation of ribonucleotides during HIV-1 reverse transcription and their attenuated repair in macrophages. J. Biol. Chem. 2012, 287, 14280–14288. [Google Scholar] [CrossRef] [Green Version]
- Goetze, R.W.; Kim, D.H.; Schinazi, R.F.; Kim, B. A CRISPR/Cas9 approach reveals that the polymerase activity of DNA polymerase beta is dispensable for HIV-1 infection in dividing and nondividing cells. J. Biol. Chem. 2017, 292, 14016–14025. [Google Scholar] [CrossRef] [Green Version]
- Van Cor-Hosmer, S.K.; Kim, D.H.; Daly, M.B.; Daddacha, W.; Kim, B. Restricted 5′-end gap repair of HIV-1 integration due to limited cellular dNTP concentrations in human primary macrophages. J. Biol. Chem. 2013, 288, 33253–33262. [Google Scholar] [CrossRef] [Green Version]
- Mahboubi, B.; Gavegnano, C.; Kim, D.H.; Schinazi, R.F.; Kim, B. Host SAMHD1 protein restricts endogenous reverse transcription of HIV-1 in nondividing macrophages. Retrovirology 2018, 15, 69. [Google Scholar] [CrossRef] [Green Version]
- Baldauf, H.M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Ambiel, I.; Wabnitz, G.; Gramberg, T.; et al. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat. Med. 2012, 18, 1682–1687. [Google Scholar] [CrossRef] [Green Version]
- Descours, B.; Cribier, A.; Chable-Bessia, C.; Ayinde, D.; Rice, G.; Crow, Y.; Yatim, A.; Schwartz, O.; Laguette, N.; Benkirane, M. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology 2012, 9, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St Gelais, C.; de Silva, S.; Amie, S.M.; Coleman, C.M.; Hoy, H.; Hollenbaugh, J.A.; Kim, B.; Wu, L. SAMHD1 restricts HIV-1 infection in dendritic cells (DCs) by dNTP depletion, but its expression in DCs and primary CD4+ T-lymphocytes cannot be upregulated by interferons. Retrovirology 2012, 9, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigdomènech, I.; Casartelli, N.; Porrot, F.; Schwartz, O. SAMHD1 restricts HIV-1 cell-to-cell transmission and limits immune detection in monocyte-derived dendritic cells. J. Virol. 2013, 87, 2846–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonucci, J.M.; Kim, S.H.; St Gelais, C.; Bonifati, S.; Li, T.W.; Buzovetsky, O.; Knecht, K.M.; Duchon, A.A.; Xiong, Y.; Musier-Forsyth, K.; et al. SAMHD1 Impairs HIV-1 Gene Expression and Negatively Modulates Reactivation of Viral Latency in CD4(+) T Cells. J. Virol. 2018, 92, e00292-18. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.H.; Bhattacharya, A.; Persaud, M.; Taylor, A.B.; Wang, Z.; Bulnes-Ramos, A.; Xu, J.; Selyutina, A.; Martinez-Lopez, A.; Cano, K.; et al. Nucleic acid binding by SAMHD1 contributes to the antiretroviral activity and is enhanced by the GpsN modification. Nat. Commun. 2021, 12, 731. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [Green Version]
- Schwefel, D.; Groom, H.C.; Boucherit, V.C.; Christodoulou, E.; Walker, P.A.; Stoye, J.P.; Bishop, K.N.; Taylor, I.A. Structural basis of lentiviral subversion of a cellular protein degradation pathway. Nature 2014, 505, 234–238. [Google Scholar] [CrossRef] [Green Version]
- Hrecka, K.; Gierszewska, M.; Srivastava, S.; Kozaczkiewicz, L.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc. Natl. Acad. Sci. USA 2007, 104, 11778–11783. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Hao, C.; Yan, J.; DeLucia, M.; Mehrens, J.; Wang, C.; Gronenborn, A.M.; Skowronski, J. HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J. Biol. Chem. 2012, 287, 12550–12558. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Swanson, S.K.; Manel, N.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 2008, 4, e1000059. [Google Scholar] [CrossRef]
- Guo, H.; Zhang, N.; Shen, S.; Yu, X.F.; Wei, W. Determinants of lentiviral Vpx-CRL4 E3 ligase-mediated SAMHD1 degradation in the substrate adaptor protein DCAF1. Biochem. Biophys. Res. Commun. 2019, 513, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Oo, A.; Kim, D.H.; Schinazi, R.F.; Kim, B. Viral protein X reduces the incorporation of mutagenic noncanonical rNTPs during lentivirus reverse transcription in macrophages. J. Biol. Chem. 2020, 295, 657–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Bailes, E.; Stevenson, M.; Emerman, M.; Hahn, B.H. Gene acquisition in HIV and SIV. Nature 1996, 383, 586–587. [Google Scholar] [CrossRef] [PubMed]
- Etienne, L.; Hahn, B.H.; Sharp, P.M.; Matsen, F.A.; Emerman, M. Gene loss and adaptation to hominids underlie the ancient origin of HIV-1. Cell Host Microbe 2013, 14, 85–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tristem, M.; Marshall, C.; Karpas, A.; Hill, F. Evolution of the primate lentiviruses: Evidence from vpx and vpr. EMBO J. 1992, 11, 3405–3412. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.S.; Fregoso, O.I.; McCoy, C.O.; Matsen, F.A.; Malik, H.S.; Emerman, M. The ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein Vpx. Cell Host Microbe 2012, 11, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Belzile, J.P.; Duisit, G.; Rougeau, N.; Mercier, J.; Finzi, A.; Cohen, E.A. HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog. 2007, 3, e85. [Google Scholar] [CrossRef] [Green Version]
- Emerman, M.; Malik, H.S. Paleovirology--modern consequences of ancient viruses. PLoS Biol. 2010, 8, e1000301. [Google Scholar] [CrossRef]
- Kirchhoff, F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 2010, 8, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Daugherty, M.D.; Malik, H.S. Rules of engagement: Molecular insights from host-virus arms races. Annu. Rev. Genet. 2012, 46, 677–700. [Google Scholar] [CrossRef]
- McNatt, M.W.; Zang, T.; Hatziioannou, T.; Bartlett, M.; Fofana, I.B.; Johnson, W.E.; Neil, S.J.; Bieniasz, P.D. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 2009, 5, e1000300. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Wu, L.I.; Emerman, M.; Malik, H.S. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. USA 2005, 102, 2832–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laguette, N.; Rahm, N.; Sobhian, B.; Chable-Bessia, C.; Münch, J.; Snoeck, J.; Sauter, D.; Switzer, W.M.; Heneine, W.; Kirchhoff, F.; et al. Evolutionary and functional analyses of the interaction between the myeloid restriction factor SAMHD1 and the lentiviral Vpx protein. Cell Host Microbe 2012, 11, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fregoso, O.I.; Ahn, J.; Wang, C.; Mehrens, J.; Skowronski, J.; Emerman, M. Evolutionary toggling of Vpx/Vpr specificity results in divergent recognition of the restriction factor SAMHD1. PLoS Pathog. 2013, 9, e1003496. [Google Scholar] [CrossRef] [Green Version]
- Lenzi, G.M.; Domaoal, R.A.; Kim, D.H.; Schinazi, R.F.; Kim, B. Mechanistic and Kinetic Differences between Reverse Transcriptases of Vpx Coding and Non-coding Lentiviruses. J. Biol. Chem. 2015, 290, 30078–30086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coggins, S.A.; Holler, J.M.; Kimata, J.T.; Kim, D.H.; Schinazi, R.F.; Kim, B. Efficient pre-catalytic conformational change of reverse transcriptases from SAMHD1 non-counteracting primate lentiviruses during dNTP incorporation. Virology 2019, 537, 36–44. [Google Scholar] [CrossRef]
- Coggins, S.A.; Kim, D.H.; Schinazi, R.F.; Desrosiers, R.C.; Kim, B. Enhanced enzyme kinetics of reverse transcriptase variants cloned from animals infected with SIVmac239 lacking Viral Protein X. J. Biol. Chem. 2020, 295, 16975–16986. [Google Scholar] [CrossRef]
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 2015, 167, 296–312. [Google Scholar] [CrossRef] [Green Version]
- Aicardi, J.; Goutieres, F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann. Neurol. 1984, 15, 49–54. [Google Scholar] [CrossRef]
- Rice, G.I.; Bond, J.; Asipu, A.; Brunette, R.L.; Manfield, I.W.; Carr, I.M.; Fuller, J.C.; Jackson, R.M.; Lamb, T.; Briggs, T.A.; et al. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet. 2009, 41, 829–832. [Google Scholar] [CrossRef] [Green Version]
- Crow, Y.J.; Hayward, B.E.; Parmar, R.; Robins, P.; Leitch, A.; Ali, M.; Black, D.N.; van Bokhoven, H.; Brunner, H.G.; Hamel, B.C.; et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet. 2006, 38, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Fye, J.M.; Orebaugh, C.D.; Coffin, S.R.; Hollis, T.; Perrino, F.W. Dominant mutation of the TREX1 exonuclease gene in lupus and Aicardi-Goutieres syndrome. J. Biol. Chem. 2011, 286, 32373–32382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orebaugh, C.D.; Fye, J.M.; Harvey, S.; Hollis, T.; Perrino, F.W. The TREX1 exonuclease R114H mutation in Aicardi-Goutières syndrome and lupus reveals dimeric structure requirements for DNA degradation activity. J. Biol. Chem. 2011, 286, 40246–40254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, Y.J.; Leitch, A.; Hayward, B.E.; Garner, A.; Parmar, R.; Griffith, E.; Ali, M.; Semple, C.; Aicardi, J.; Babul-Hirji, R.; et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat. Genet. 2006, 38, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lopez, A.; Martin-Fernandez, M.; Buta, S.; Kim, B.; Bogunovic, D.; Diaz-Griffero, F. SAMHD1 deficient human monocytes autonomously trigger type I interferon. Mol. Immunol. 2018, 101, 450–460. [Google Scholar] [CrossRef]
- Seamon, K.J.; Sun, Z.; Shlyakhtenko, L.S.; Lyubchenko, Y.L.; Stivers, J.T. SAMHD1 is a single-stranded nucleic acid binding protein with no active site-associated nuclease activity. Nucleic Acids Res. 2015, 43, 6486–6499. [Google Scholar] [CrossRef] [Green Version]
- Seamon, K.J.; Bumpus, N.N.; Stivers, J.T. Single-Stranded Nucleic Acids Bind to the Tetramer Interface of SAMHD1 and Prevent Formation of the Catalytic Homotetramer. Biochemistry 2016, 55, 6087–6099. [Google Scholar] [CrossRef] [Green Version]
- Maelfait, J.; Bridgeman, A.; Benlahrech, A.; Cursi, C.; Rehwinkel, J. Restriction by SAMHD1 Limits cGAS/STING-Dependent Innate and Adaptive Immune Responses to HIV-1. Cell Rep. 2016, 16, 1492–1501. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, A.; Karayel, E.; Rice, G.I.; Bennett, K.L.; Crow, Y.J.; Superti-Furga, G.; Bürckstümmer, T. SAMHD1 is a nucleic-acid binding protein that is mislocalized due to aicardi-goutières syndrome-associated mutations. Hum. Mutat. 2012, 33, 1116–1122. [Google Scholar] [CrossRef]
- Chen, S.; Bonifati, S.; Qin, Z.; St Gelais, C.; Kodigepalli, K.M.; Barrett, B.S.; Kim, S.H.; Antonucci, J.M.; Ladner, K.J.; Buzovetsky, O.; et al. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-kappaB and interferon pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E3798–E3807. [Google Scholar] [PubMed] [Green Version]
- Chen, S.; Bonifati, S.; Qin, Z.; St Gelais, C.; Wu, L. SAMHD1 Suppression of Antiviral Immune Responses. Trends Microbiol. 2019, 27, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Bonifati, S.; St Gelais, C.; Li, T.W.; Kim, S.H.; Antonucci, J.M.; Mahboubi, B.; Yount, J.S.; Xiong, Y.; Kim, B.; et al. The dNTPase activity of SAMHD1 is important for its suppression of innate immune responses in differentiated monocytic cells. J. Biol. Chem. 2020, 295, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Espada, C.E.; St Gelais, C.; Bonifati, S.; Maksimova, V.V.; Cahill, M.P.; Kim, S.H.; Wu, L. TRAF6 and TAK1 Contribute to SAMHD1-Mediated Negative Regulation of NF-κB Signaling. J. Virol. 2021, 95, e01970-20. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Langer, S.; Zhang, Z.; Herbert, K.M.; Yoh, S.; König, R.; Chanda, S.K. Sensor Sensibility-HIV-1 and the Innate Immune Response. Cells 2020, 9, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; de Silva, S.; Wang, J.H.; Wu, L. Co-evolution of primate SAMHD1 and lentivirus Vpx leads to the loss of the vpx gene in HIV-1 ancestor. PLoS ONE 2012, 7, e37477. [Google Scholar] [CrossRef]
- Oo, A.; Zandi, K.; Shepard, C.; Bassit, L.C.; Musall, K.; Goh, S.L.; Cho, Y.J.; Kim, D.H.; Schinazi, R.F.; Kim, B. Elimination of Aicardi-Goutières syndrome protein SAMHD1 activates cellular innate immunity and suppresses SARS-CoV-2 replication. J. Biol. Chem. 2022, 298, 101635. [Google Scholar] [CrossRef]
- Mohamed, A.; Bakir, T.; Al-Hawel, H.; Al-Sharif, I.; Bakheet, R.; Kouser, L.; Murugaiah, V.; Al-Mozaini, M. HIV-2 Vpx neutralizes host restriction factor SAMHD1 to promote viral pathogenesis. Sci. Rep. 2021, 11, 20984. [Google Scholar] [CrossRef]
- Fink, D.L.; Cai, J.; Whelan, M.V.X.; Monit, C.; Maluquer de Motes, C.; Towers, G.J.; Sumner, R.P. HIV-2/SIV Vpx antagonises NF-κB activation by targeting p65. Retrovirology 2022, 19, 2. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, M.; Pan, X.; Zhu, Y.; Yan, H.; Jiang, T.; Shen, Y.; Dong, X.; Zheng, N.; Lu, J.; et al. Inhibition of Hepatitis B virus replication by SAMHD1. Biochem. Biophys. Res. Commun. 2014, 450, 1462–1468. [Google Scholar] [CrossRef]
- Cingöz, O.; Arnow, N.D.; Puig Torrents, M.; Bannert, N. Vpx enhances innate immune responses independently of SAMHD1 during HIV-1 infection. Retrovirology 2021, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Meuth, M.; L’Heureux-Huard, N.; Trudel, M. Characterization of a mutator gene in Chinese hamster ovary cells. Proc. Natl. Acad. Sci. USA 1979, 76, 6505–6509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, G.; Ullman, B.; Martin, D.W., Jr. Mutator phenotypes in mammalian cell mutants with distinct biochemical defects and abnormal deoxyribonucleoside triphosphate pools. Proc. Natl. Acad. Sci. USA 1981, 78, 2447–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkel, T.A. DNA replication fidelity. J. Biol. Chem. 1992, 267, 18251–18254. [Google Scholar] [CrossRef]
- Kumar, D.; Abdulovic, A.L.; Viberg, J.; Nilsson, A.K.; Kunkel, T.A.; Chabes, A. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 2011, 39, 1360–1371. [Google Scholar] [CrossRef] [Green Version]
- Traut, T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef]
- Amin, N.A.; Seymour, E.; Saiya-Cork, K.; Parkin, B.; Shedden, K.; Malek, S.N. A Quantitative Analysis of Subclonal and Clonal Gene Mutations before and after Therapy in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2016, 22, 4525–4535. [Google Scholar] [CrossRef] [Green Version]
- Clifford, R.; Louis, T.; Robbe, P.; Ackroyd, S.; Burns, A.; Timbs, A.T.; Wright Colopy, G.; Dreau, H.; Sigaux, F.; Judde, J.G.; et al. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood 2014, 123, 1021–1031. [Google Scholar] [CrossRef]
- Johansson, P.; Klein-Hitpass, L.; Choidas, A.; Habenberger, P.; Mahboubi, B.; Kim, B.; Bergmann, A.; Scholtysik, R.; Brauser, M.; Lollies, A.; et al. SAMHD1 is recurrently mutated in T-cell prolymphocytic leukemia. Blood Cancer J. 2018, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Li, C.; Liu, Z.; Shengjing, H. Expression and Relationship of SAMHD1 with Other Apoptotic and Autophagic Genes in Acute Myeloid Leukemia Patients. Acta Haematol. 2020, 143, 51–59. [Google Scholar] [CrossRef]
- Guièze, R.; Robbe, P.; Clifford, R.; de Guibert, S.; Pereira, B.; Timbs, A.; Dilhuydy, M.S.; Cabes, M.; Ysebaert, L.; Burns, A.; et al. Presence of multiple recurrent mutations confers poor trial outcome of relapsed/refractory CLL. Blood 2015, 126, 2110–2117. [Google Scholar] [CrossRef] [Green Version]
- de Silva, S.; Wang, F.; Hake, T.S.; Porcu, P.; Wong, H.K.; Wu, L. Downregulation of SAMHD1 expression correlates with promoter DNA methylation in Sézary syndrome patients. J. Investig. Dermatol. 2014, 134, 562–565. [Google Scholar] [CrossRef] [Green Version]
- Merati, M.; Buethe, D.J.; Cooper, K.D.; Honda, K.S.; Wang, H.; Gerstenblith, M.R. Aggressive CD8(+) epidermotropic cutaneous T-cell lymphoma associated with homozygous mutation in SAMHD1. JAAD Case Rep. 2015, 1, 227–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.L.; Lu, F.Z.; Shen, X.Y.; Wu, Y.; Zhao, L.T. SAMHD1 is down regulated in lung cancer by methylation and inhibits tumor cell proliferation. Biochem. Biophys. Res. Commun. 2014, 455, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Rentoft, M.; Lindell, K.; Tran, P.; Chabes, A.L.; Buckland, R.J.; Watt, D.L.; Marjavaara, L.; Nilsson, A.K.; Melin, B.; Trygg, J.; et al. Heterozygous colon cancer-associated mutations of SAMHD1 have functional significance. Proc. Natl. Acad. Sci. USA 2016, 113, 4723–4728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef]
- Bowen, N.E.; Temple, J.; Shepard, C.; Oo, A.; Arizaga, F.; Kapoor-Vazirani, P.; Persaud, M.; Yu, C.H.; Kim, D.H.; Schinazi, R.F.; et al. Structural and functional characterization explains loss of dNTPase activity of the cancer-specific R366C/H mutant SAMHD1 proteins. J. Biol. Chem. 2021, 297, 101170. [Google Scholar] [CrossRef]
- Herold, N.; Rudd, S.G.; Sanjiv, K.; Kutzner, J.; Myrberg, I.H.; Paulin, C.B.J.; Olsen, T.K.; Helleday, T.; Henter, J.I.; Schaller, T. With me or against me: Tumor suppressor and drug resistance activities of SAMHD1. Exp. Hematol. 2017, 52, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Knecht, K.M.; Buzovetsky, O.; Schneider, C.; Thomas, D.; Srikanth, V.; Kaderali, L.; Tofoleanu, F.; Reiss, K.; Ferreiros, N.; Geisslinger, G.; et al. The structural basis for cancer drug interactions with the catalytic and allosteric sites of SAMHD1. Proc. Natl. Acad. Sci. USA 2018, 115, E10022–E10031. [Google Scholar] [CrossRef] [Green Version]
- Herold, N.; Rudd, S.G.; Sanjiv, K.; Kutzner, J.; Bladh, J.; Paulin, C.B.J.; Helleday, T.; Henter, J.I.; Schaller, T. SAMHD1 protects cancer cells from various nucleoside-based antimetabolites. Cell Cycle 2017, 16, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Rothenburger, T.; McLaughlin, K.M.; Herold, T.; Schneider, C.; Oellerich, T.; Rothweiler, F.; Feber, A.; Fenton, T.R.; Wass, M.N.; Keppler, O.T.; et al. SAMHD1 is a key regulator of the lineage-specific response of acute lymphoblastic leukaemias to nelarabine. Commun. Biol. 2020, 3, 324. [Google Scholar] [CrossRef] [PubMed]
- Rothenburger, T.; Thomas, D.; Schreiber, Y.; Wratil, P.R.; Pflantz, T.; Knecht, K.; Digianantonio, K.; Temple, J.; Schneider, C.; Baldauf, H.M.; et al. Differences between intrinsic and acquired nucleoside analogue resistance in acute myeloid leukaemia cells. J. Exp. Clin. Cancer Res. 2021, 40, 317. [Google Scholar] [CrossRef] [PubMed]
- Rudd, S.G.; Tsesmetzis, N.; Sanjiv, K.; Paulin, C.B.; Sandhow, L.; Kutzner, J.; Hed Myrberg, I.; Bunten, S.S.; Axelsson, H.; Zhang, S.M.; et al. Ribonucleotide reductase inhibitors suppress SAMHD1 ara-CTPase activity enhancing cytarabine efficacy. EMBO Mol. Med. 2020, 12, e10419. [Google Scholar] [CrossRef]
- Daddacha, W.; Koyen, A.E.; Bastien, A.J.; Head, P.E.; Dhere, V.R.; Nabeta, G.N.; Connolly, E.C.; Werner, E.; Madden, M.Z.; Daly, M.B.; et al. SAMHD1 Promotes DNA End Resection to Facilitate DNA Repair by Homologous Recombination. Cell Rep. 2017, 20, 1921–1935. [Google Scholar] [CrossRef] [Green Version]
- Coquel, F.; Silva, M.J.; Techer, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.L.; et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Akimova, E.; Gassner, F.J.; Schubert, M.; Rebhandl, S.; Arzt, C.; Rauscher, S.; Tober, V.; Zaborsky, N.; Greil, R.; Geisberger, R. SAMHD1 restrains aberrant nucleotide insertions at repair junctions generated by DNA end joining. Nucleic Acids Res. 2021, 49, 2598–2608. [Google Scholar] [CrossRef]
- Husain, A.; Xu, J.; Fujii, H.; Nakata, M.; Kobayashi, M.; Wang, J.Y.; Rehwinkel, J.; Honjo, T.; Begum, N.A. SAMHD1-mediated dNTP degradation is required for efficient DNA repair during antibody class switch recombination. EMBO J. 2020, 39, e102931. [Google Scholar] [CrossRef]
- Park, K.; Ryoo, J.; Jeong, H.; Kim, M.; Lee, S.; Hwang, S.Y.; Ahn, J.; Kim, D.; Moon, H.C.; Baek, D.; et al. Aicardi-Goutières syndrome-associated gene SAMHD1 preserves genome integrity by preventing R-loop formation at transcription-replication conflict regions. PLoS Genet. 2021, 17, e1009523. [Google Scholar] [CrossRef]
- Franzolin, E.; Coletta, S.; Ferraro, P.; Pontarin, G.; D’Aronco, G.; Stevanoni, M.; Palumbo, E.; Cagnin, S.; Bertoldi, L.; Feltrin, E.; et al. SAMHD1-deficient fibroblasts from Aicardi-Goutieres Syndrome patients can escape senescence and accumulate mutations. FASEB J. 2020, 34, 631–647. [Google Scholar] [CrossRef] [Green Version]
- Kretschmer, S.; Wolf, C.; Konig, N.; Staroske, W.; Guck, J.; Hausler, M.; Luksch, H.; Nguyen, L.A.; Kim, B.; Alexopoulou, D.; et al. SAMHD1 prevents autoimmunity by maintaining genome stability. Ann. Rheum. Dis. 2015, 74, e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coggins, S.A.; Mahboubi, B.; Schinazi, R.F.; Kim, B. SAMHD1 Functions and Human Diseases. Viruses 2020, 12, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Zyl, G.; Bale, M.J.; Kearney, M.F. HIV evolution and diversity in ART-treated patients. Retrovirology 2018, 15, 14. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Sobieszczyk, M.E.; McCutchan, F.E.; Hammer, S.M. The challenge of HIV-1 subtype diversity. N. Engl. J. Med. 2008, 358, 1590–1602. [Google Scholar] [CrossRef] [Green Version]
- Back, N.K.; Nijhuis, M.; Keulen, W.; Boucher, C.A.; Oude Essink, B.O.; van Kuilenburg, A.B.; van Gennip, A.H.; Berkhout, B. Reduced replication of 3TC-resistant HIV-1 variants in primary cells due to a processivity defect of the reverse transcriptase enzyme. EMBO J. 1996, 15, 4040–4049. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, M.M.; Karvonen, S.C.; Gaba, A.; Flath, B.; Chelico, L.; Emerman, M. Highly-potent, synthetic APOBEC3s restrict HIV-1 through deamination-independent mechanisms. PLoS Pathog. 2021, 17, e1009523. [Google Scholar] [CrossRef]
- Sadler, H.A.; Stenglein, M.D.; Harris, R.S.; Mansky, L.M. APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. J. Virol. 2010, 84, 7396–7404. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowen, N.E.; Oo, A.; Kim, B. Mechanistic Interplay between HIV-1 Reverse Transcriptase Enzyme Kinetics and Host SAMHD1 Protein: Viral Myeloid-Cell Tropism and Genomic Mutagenesis. Viruses 2022, 14, 1622. https://doi.org/10.3390/v14081622
Bowen NE, Oo A, Kim B. Mechanistic Interplay between HIV-1 Reverse Transcriptase Enzyme Kinetics and Host SAMHD1 Protein: Viral Myeloid-Cell Tropism and Genomic Mutagenesis. Viruses. 2022; 14(8):1622. https://doi.org/10.3390/v14081622
Chicago/Turabian StyleBowen, Nicole E., Adrian Oo, and Baek Kim. 2022. "Mechanistic Interplay between HIV-1 Reverse Transcriptase Enzyme Kinetics and Host SAMHD1 Protein: Viral Myeloid-Cell Tropism and Genomic Mutagenesis" Viruses 14, no. 8: 1622. https://doi.org/10.3390/v14081622