
Comparative Dynamics of Delta and Omicron SARS-CoV-2 Variants across and between California and Mexico


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Compilation of Datasets and Data Preparation for Phylogenetic Inference
2.2. Subsampling
2.3. Lineage Identification
2.4. Identification of Representative Phylogenetic Clades
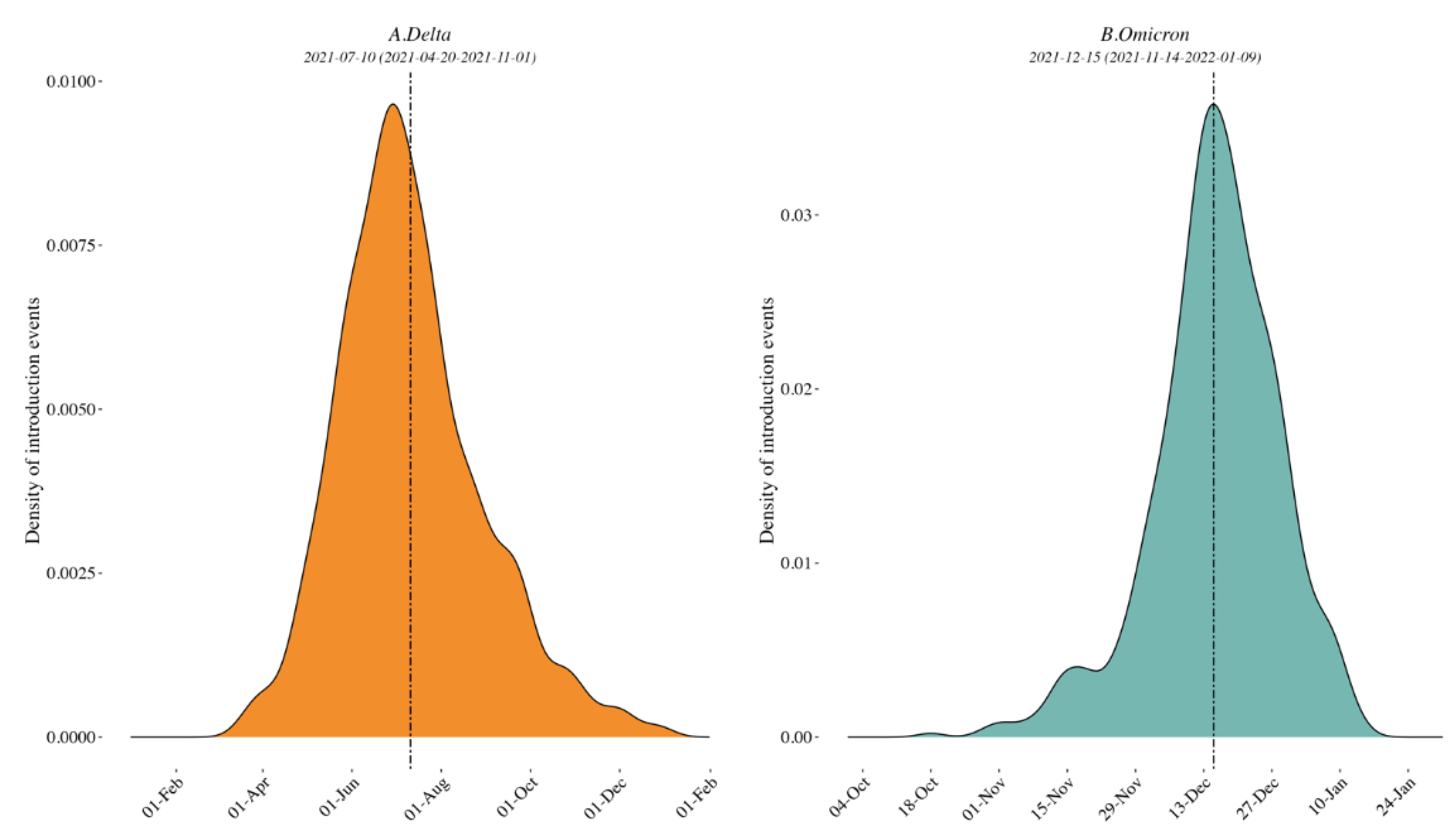
2.5. Timing of External Introductions
2.6. Phylogeographic Inference
2.7. Mobility Data
3. Results
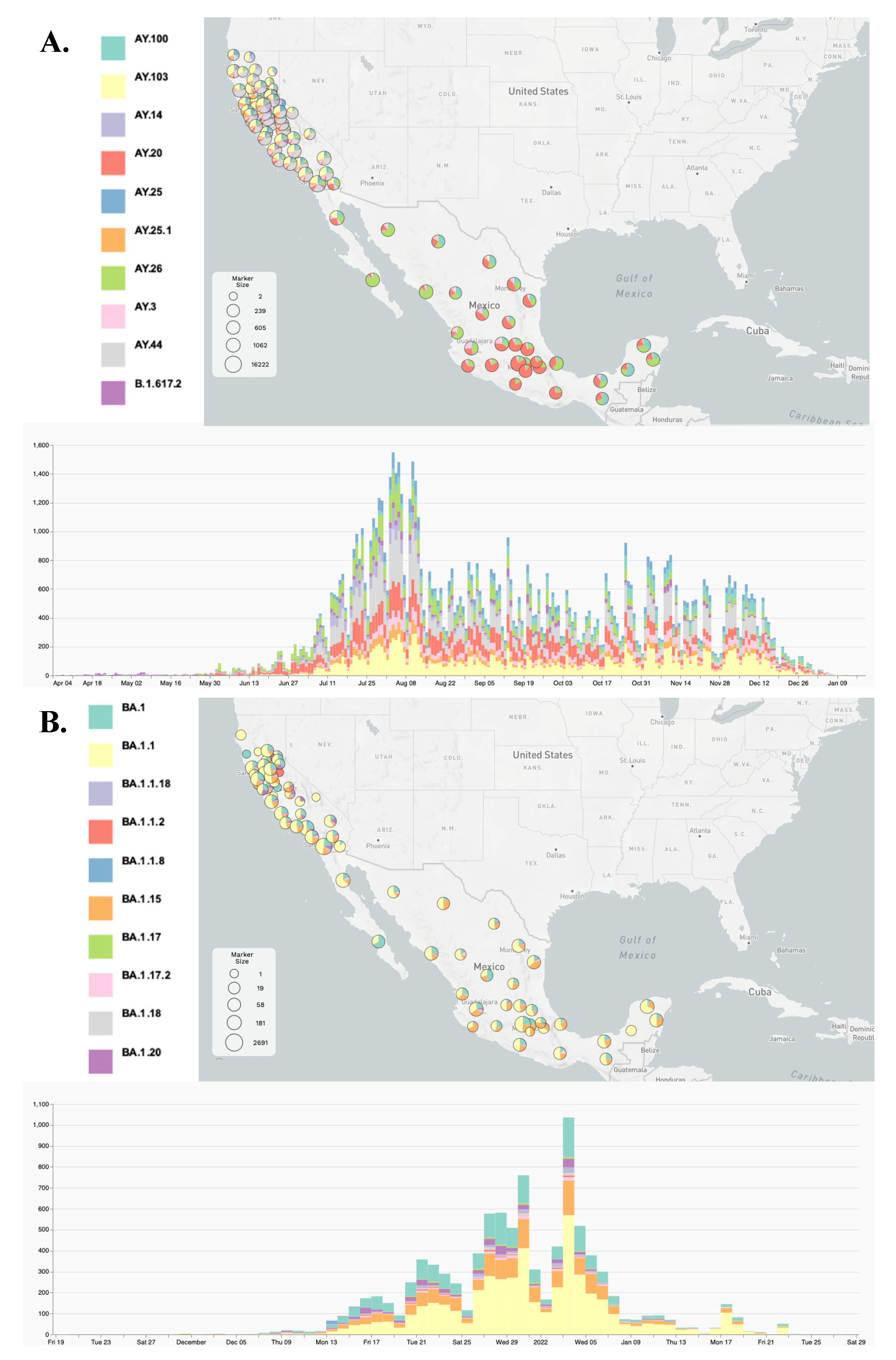
3.1. Clade Identification and Variant External Introduction in the Border Region
3.2. Lineage Distribution of the Delta and Omicron Variants
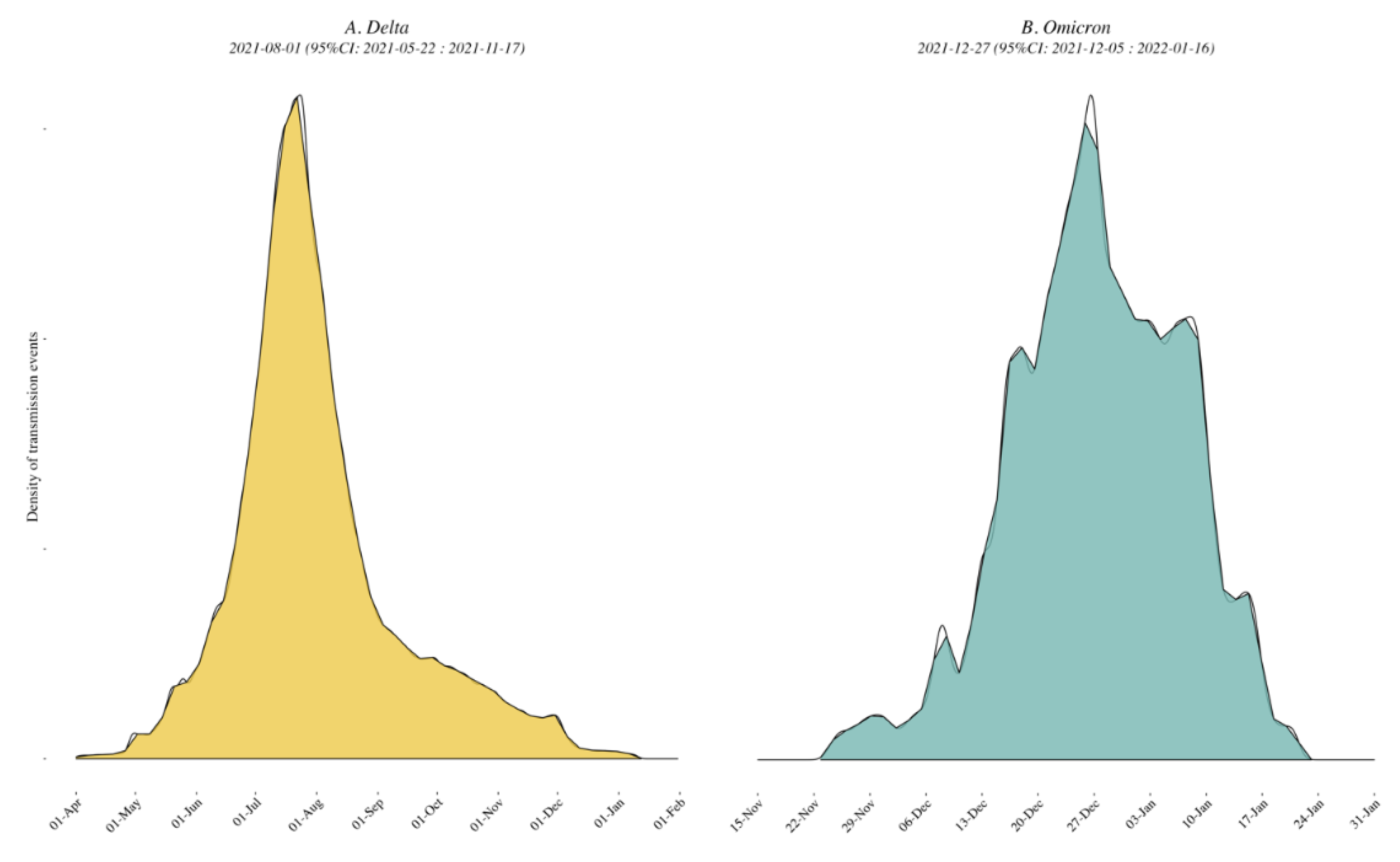
3.3. Viral Dynamics within California and Mexico
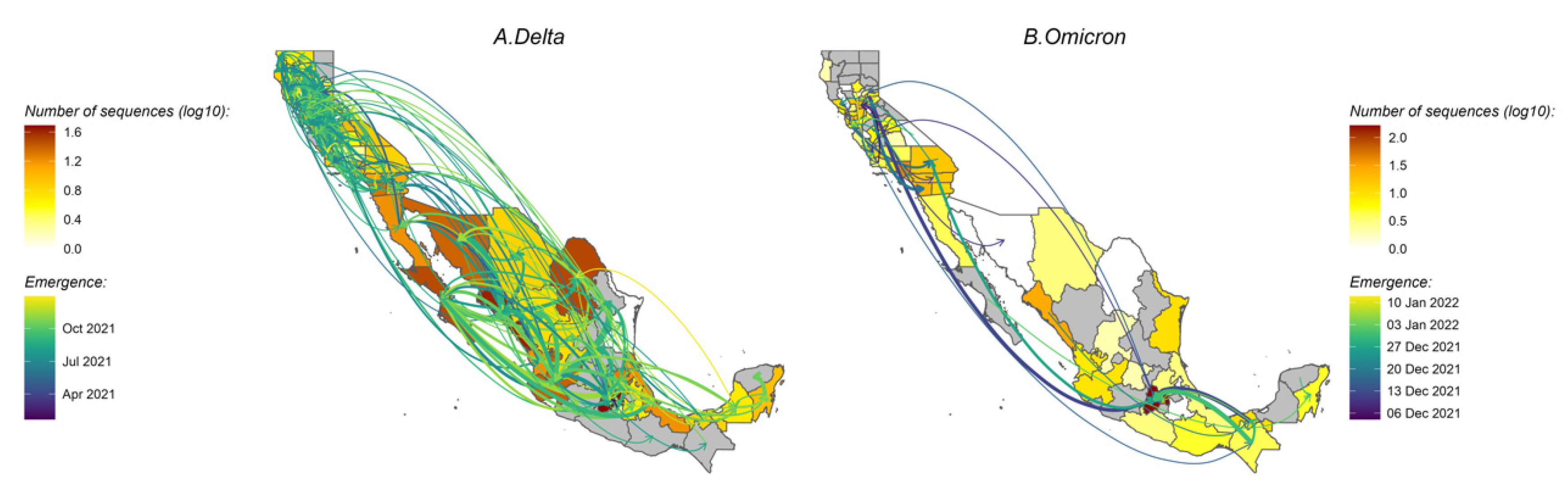
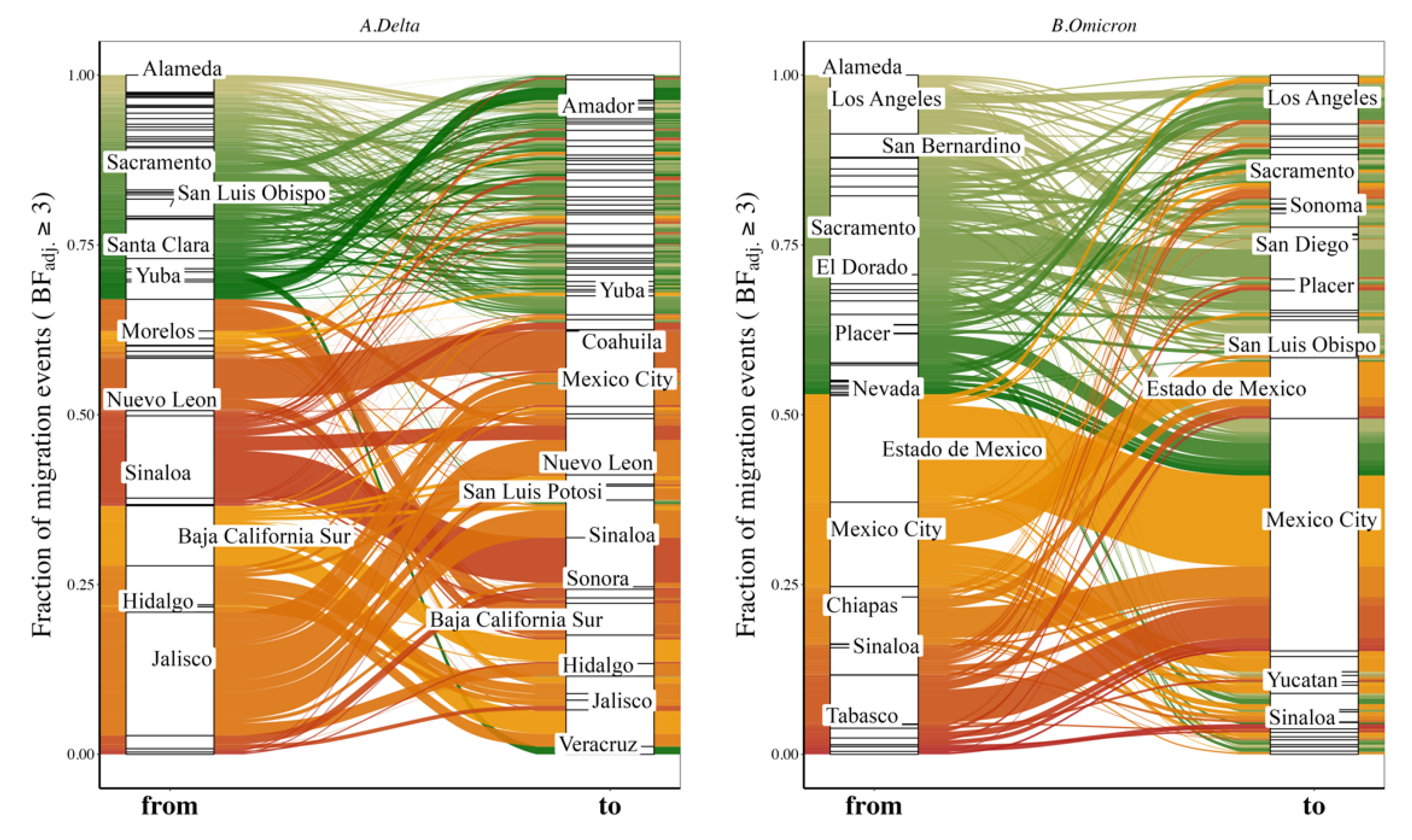
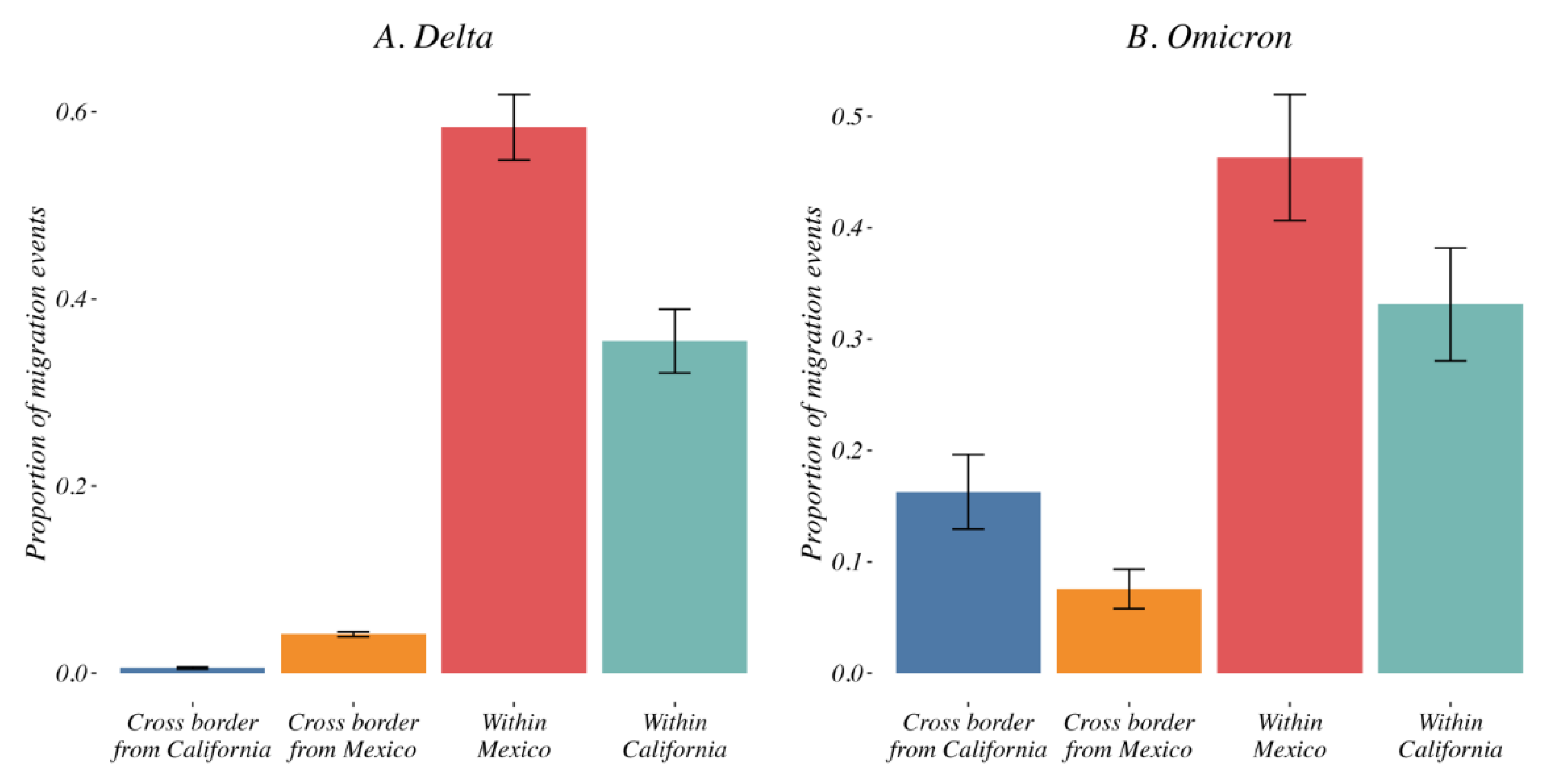
3.4. Variant Migration across the California–Mexico Border
3.5. Human Mobility and Variant Dispersal
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; du Plessis, L.; Liu, Z.; Hill, V.; Kang, M.; Lin, H.; Sun, J.; François, S.; Kraemer, M.U.G.; Faria, N.R.; et al. Genomic Epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell 2020, 181, 997–1003.e9. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. Alarming COVID variants show vital role of genomic surveillance. Nature 2021, 589, 337–338. [Google Scholar] [CrossRef]
- Shepardson, D. U.S. Extends Travel Curbs at Canada, Mexico Land Borders through 21 August. Available online: https://www.reuters.com/world/us/us-extends-travel-restrictions-canada-mexico-land-borders-through-aug-21-2021-07-21/ (accessed on 15 February 2022).
- Lambrou, A.S.; Shirk, P.; Steele, M.K.; Paul, P.; Paden, C.R.; Cadwell, B.; Reese, H.E.; Aoki, Y.; Hassell, N.; Zheng, X.Y.; et al. Genomic Surveillance for SARS-CoV-2 Variants: Predominance of the Delta (B.1.617.2) and Omicron (B.1.1.529) Variants—United States, June 2021–January 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 206–211. [Google Scholar] [CrossRef]
- Nishiura, H.; Ito, K.; Anzai, A.; Kobayashi, T.; Piantham, C.; Rodriguez-Morales, A.J. Relative Reproduction Number of SARS-CoV-2 Omicron (B.1.1.529) Compared with Delta Variant in South Africa. J. Clin. Med. 2021, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Du Plessis, L.; McCrone, J.T.; Zarebski, A.E.; Hill, V.; Ruis, C.; Gutierrez, B.; Raghwani, J.; Ashworth, J.; Colquhoun, R.; Connor, T.R.; et al. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 2021, 371, 708–712. [Google Scholar] [CrossRef]
- Dellicour, S.; Hong, S.L.; Vrancken, B.; Chaillon, A.; Gill, M.S.; Maurano, M.T.; Ramaswami, S.; Zappile, P.; Marier, C.; Harkins, G.W.; et al. Dispersal dynamics of SARS-CoV-2 lineages during the first epidemic wave in New York City. PLoS Pathog. 2021, 17, e1009571. [Google Scholar] [CrossRef]
- Dellicour, S.; Durkin, K.; Hong, S.L.; Vanmechelen, B.; Martí-Carreras, J.; Gill, M.S.; Meex, C.; Bontems, S.; André, E.; Gilbert, M.; et al. A Phylodynamic Workflow to Rapidly Gain Insights into the Dispersal History and Dynamics of SARS-CoV-2 Lineages. Mol. Biol. Evol. 2021, 38, 1608–1613. [Google Scholar] [CrossRef]
- Kraemer, M.U.G.; Hill, V.; Ruis, C.; Dellicour, S.; Bajaj, S.; McCrone, J.T.; Baele, G.; Parag, K.V.; Battle, A.L.; Gutierrez, B.; et al. Spatiotemporal invasion dynamics of SARS-CoV-2 lineage B.1.1.7 emergence. Science 2021, 373, 889–895. [Google Scholar] [CrossRef]
- Rambaut, A.; Pybus, O.G.; Nelson, M.I.; Viboud, C.; Taubenberger, J.K.; Holmes, E.C. The genomic and epidemiological dynamics of human influenza A virus. Nature 2008, 453, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eur. Surveill. Eur. Commun. Dis. Bull. 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- United States Department of Transportation Bureau of Transportaion Statistics (BTS). Available online: https://www.bts.gov/topics/national-transportation-statistics (accessed on 1 March 2022).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Argimón, S.; Abudahab, K.; Goater, R.J.E.; Fedosejev, A.; Bhai, J.; Glasner, C.; Feil, E.J.; Holden, M.T.G.; Yeats, C.A.; Grundmann, H.; et al. Microreact: Visualizing and sharing data for genomic epidemiology and phylogeography. Microb. Genom. 2016, 2, e000093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- USAFacts Understanding the COVID-19 Pandemic. Available online: https://usafacts.org/issues/coronavirus/ (accessed on 13 February 2022).
- Government of Mexico COVID-19 Data for Mexico. Available online: https://datos.covid-19.conacyt.mx/#DOView (accessed on 13 February 2022).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, L.; Vrancken, B.; Fabeni, L.; Marascio, N.; Cento, V.; Di Maio, V.C.; Aragri, M.; Pineda-Pena, A.C.; Schrooten, Y.; Van Laethem, K.; et al. Implications of hepatitis C virus subtype 1a migration patterns for virus genetic sequencing policies in Italy. BMC Evol. Biol. 2017, 17, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.M.; Frost, S.D.W. Scalable relaxed clock phylogenetic dating. Virus Evol. 2017, 3, vex025. [Google Scholar] [CrossRef] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Minin, V.N.; Bloomquist, E.W.; Suchard, M.A. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol. Biol. Evol. 2008, 25, 1459–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaillon, A.; Gianella, S.; Dellicour, S.; Rawlings, S.A.; Schlub, T.E.; Faria De Oliveira, M.; Ignacio, C.; Porrachia, M.; Vrancken, B.; Smith, D.M. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. J. Clin. Investig. 2020, 130, 1699–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kass, R.E.; Raftery, A.E. Bayes Factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Maas, P. Facebook Disaster Maps: Aggregate Insights for Crisis Response & Recovery. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 4–8 August 2019; p. 3173. [Google Scholar]
- Lemey, P.; Ruktanonchai, N.; Hong, S.L.; Colizza, V.; Poletto, C.; Van den Broeck, F.; Gill, M.S.; Ji, X.; Levasseur, A.; Oude Munnink, B.B.; et al. Untangling introductions and persistence in COVID-19 resurgence in Europe. Nature 2021, 595, 713–717. [Google Scholar] [CrossRef]
- Deng, X.; Gu, W.; Federman, S.; du Plessis, L.; Pybus, O.G.; Faria, N.; Wang, C.; Yu, G.; Pan, C.-Y.; Guevara, H.; et al. A Genomic Survey of SARS-CoV-2 Reveals Multiple Introductions into Northern California without a Predominant Lineage. Science 2020, 369, 582–587. [Google Scholar] [CrossRef]
- Annavajhala, M.K.; Mohri, H.; Wang, P.; Nair, M.; Zucker, J.E.; Sheng, Z.; Gomez-Simmonds, A.; Kelley, A.L.; Tagliavia, M.; Huang, Y.; et al. Emergence and expansion of SARS-CoV-2 B.1.526 after identification in New York. Nature 2021, 597, 703–708. [Google Scholar] [CrossRef]
- Kumar, S.; Thambiraja, T.S.; Karuppanan, K.; Subramaniam, G. Omicron and Delta variant of SARS-CoV-2: A comparative computational study of spike protein. J. Med. Virol. 2022, 94, 1641–1649. [Google Scholar] [CrossRef]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef]
- Shishir, T.A.; Jannat, T.; Naser, I.B. An in-silico study of the mutation-associated effects on the spike protein of SARS-CoV-2, Omicron variant. PLoS ONE 2022, 17, e0266844. [Google Scholar] [CrossRef]
- Medigeshi, G.; Batra, G.; Murugesan, D.R.; Thiruvengadam, R.; Chattopadhyay, S.; Das, B.; Gosain, M.; Ayushi; Singh, J.; Anbalagan, A.; et al. Sub-optimal Neutralisation of Omicron (B.1.1.529) Variant by Antibodies induced by Vaccine alone or SARS-CoV-2 Infection plus Vaccine (Hybrid Immunity) post 6-months. eBioMedicine 2022, 78, 103938. [Google Scholar] [CrossRef]
- Ai, J.; Zhang, H.; Zhang, Y.; Lin, K.; Zhang, Y.; Wu, J.; Wan, Y.; Huang, Y.; Song, J.; Fu, Z.; et al. Omicron variant showed lower neutralizing sensitivity than other SARS-CoV-2 variants to immune sera elicited by vaccines after boost. Emerg. Microbes Infect. 2022, 11, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, M.U.G.; Yang, C.H.; Gutierrez, B.; Wu, C.H.; Klein, B.; Pigott, D.M.; du Plessis, L.; Faria, N.R.; Li, R.; Hanage, W.P.; et al. The effect of human mobility and control measures on the COVID-19 epidemic in China. Science 2020, 368, 493–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Bedford, T.; Faria, N.; Bielejec, F.; Baele, G.; Russell, C.A.; Smith, D.J.; Pybus, O.G.; Brockmann, D. Unifying viral genetics and human transportation data to predict the global transmission dynamics of human influenza H3N2. PloS Pathog. 2014, 10, e1003932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwee, S.X.W.; Chua, P.E.Y.; Wang, M.X.; Pang, J. Impact of travel ban implementation on COVID-19 spread in Singapore, Taiwan, Hong Kong and South Korea during the early phase of the pandemic: A comparative study. BMC Infect. Dis. 2021, 21, 799. [Google Scholar] [CrossRef]
- Burns, J.; Movsisyan, A.; Stratil, J.M.; Biallas, R.L.; Coenen, M.; Emmert-Fees, K.M.; Geffert, K.; Hoffmann, S.; Horstick, O.; Laxy, M.; et al. International travel-related control measures to contain the COVID-19 pandemic: A rapid review. Cochrane Database Syst. Rev. 2021, 3, CD013717. [Google Scholar] [PubMed]
- US Homeland Security Guidance for Travelers to Enter the U.S. at Land Ports of Entry and Ferry Terminals. Available online: https://www.dhs.gov/news/2021/10/29/fact-sheet-guidance-travelers-enter-us-land-ports-entry-and-ferry-terminals (accessed on 13 February 2022).
- Diesel, J.; Sterrett, N.; Dasgupta, S.; Kriss, J.L.; Barry, V.; Vanden Esschert, K.; Whiteman, A.; Cadwell, B.L.; Weller, D.; Qualters, J.R.; et al. COVID-19 Vaccination Coverage among Adults—United States, 14 December 2020–22 May 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 922–927. [Google Scholar] [CrossRef]
- Reuters.com COVID-19 Vaccination Tracker. Available online: https://graphics.reuters.com/world-coronavirus-tracker-and-maps/vaccination-rollout-and-access/ (accessed on 19 February 2022).
- De Maio, N.; Wu, C.-H.; O’Reilly, K.M.; Wilson, D. New Routes to Phylogeography: A Bayesian Structured Coalescent Approximation. PLoS Genet. 2015, 11, e1005421. [Google Scholar] [CrossRef] [Green Version]
- Kalkauskas, A.; Perron, U.; Sun, Y.; Goldman, N.; Baele, G.; Guindon, S.; De Maio, N. Sampling bias and model choice in continuous phylogeography: Getting lost on a random walk. PLoS Comput. Biol. 2021, 17, e1008561. [Google Scholar] [CrossRef]
- Spiliotis, K.; Koutsoumaris, C.C.; Reppas, A.I.; Papaxenopoulou, L.A.; Starke, J.; Hatzikirou, H. Optimal vaccine roll-out strategies including social distancing for pandemics. iScience 2022, 25, 104575. [Google Scholar] [CrossRef]
- Beira, M.J.; Sebastião, P.J. A differential equations model-fitting analysis of COVID-19 epidemiological data to explain multi-wave dynamics. Sci. Rep. 2021, 11, 16312. [Google Scholar] [CrossRef] [PubMed]
- El Koufi, A.; El Koufi, N. Stochastic differential equation model of Covid-19: Case study of Pakistan. Results Phys. 2022, 34, 105218. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.; El-Kafrawy, S.A.; Alandijany, T.A.; Giannino, F.; Mirza, A.A.; El-Daly, M.M.; Faizo, A.A.; Bajrai, L.H.; Kamal, M.A.; Azhar, E.I. A global report on the dynamics of COVID-19 with quarantine and hospitalization: A fractional order model with non-local kernel. Comput. Biol. Chem. 2022, 98, 107645. [Google Scholar] [CrossRef] [PubMed]
- Sintunavarat, W.; Turab, A. Mathematical analysis of an extended SEIR model of COVID-19 using the ABC-fractional operator. Math. Comput. Simul. 2022, 198, 65–84. [Google Scholar] [CrossRef]
- Majee, S.; Adak, S.; Jana, S.; Mandal, M.; Kar, T.K. Complex dynamics of a fractional-order SIR system in the context of COVID-19. J. Appl. Math. Comput. 2022, 1–24. [Google Scholar] [CrossRef]
- Boonpatcharanon, S.; Heffernan, J.M.; Jankowski, H. Estimating the basic reproduction number at the beginning of an outbreak. PLoS ONE 2022, 17, e0269306. [Google Scholar] [CrossRef]
- Mwalili, S.; Kimathi, M.; Ojiambo, V.; Gathungu, D.; Mbogo, R. SEIR model for COVID-19 dynamics incorporating the environment and social distancing. BMC Res. Notes 2020, 13, 352. [Google Scholar] [CrossRef]
- Kucharski, A.J.; Klepac, P.; Conlan, A.J.K.; Kissler, S.M.; Tang, M.L.; Fry, H.; Gog, J.R.; Edmunds, W.J. Effectiveness of isolation, testing, contact tracing, and physical distancing on reducing transmission of SARS-CoV-2 in different settings: A mathematical modelling study. Lancet Infect. Dis. 2020, 20, 1151–1160. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehta, S.R.; Smith, D.M.; Boukadida, C.; Chaillon, A. Comparative Dynamics of Delta and Omicron SARS-CoV-2 Variants across and between California and Mexico. Viruses 2022, 14, 1494. https://doi.org/10.3390/v14071494
Mehta SR, Smith DM, Boukadida C, Chaillon A. Comparative Dynamics of Delta and Omicron SARS-CoV-2 Variants across and between California and Mexico. Viruses. 2022; 14(7):1494. https://doi.org/10.3390/v14071494
Chicago/Turabian StyleMehta, Sanjay R., Davey M. Smith, Celia Boukadida, and Antoine Chaillon. 2022. "Comparative Dynamics of Delta and Omicron SARS-CoV-2 Variants across and between California and Mexico" Viruses 14, no. 7: 1494. https://doi.org/10.3390/v14071494