
Understanding Immune Responses to Viruses—Do Underlying Th1/Th2 Cell Biases Predict Outcome?

 , ,
, ,
Abstract
:1. Introduction
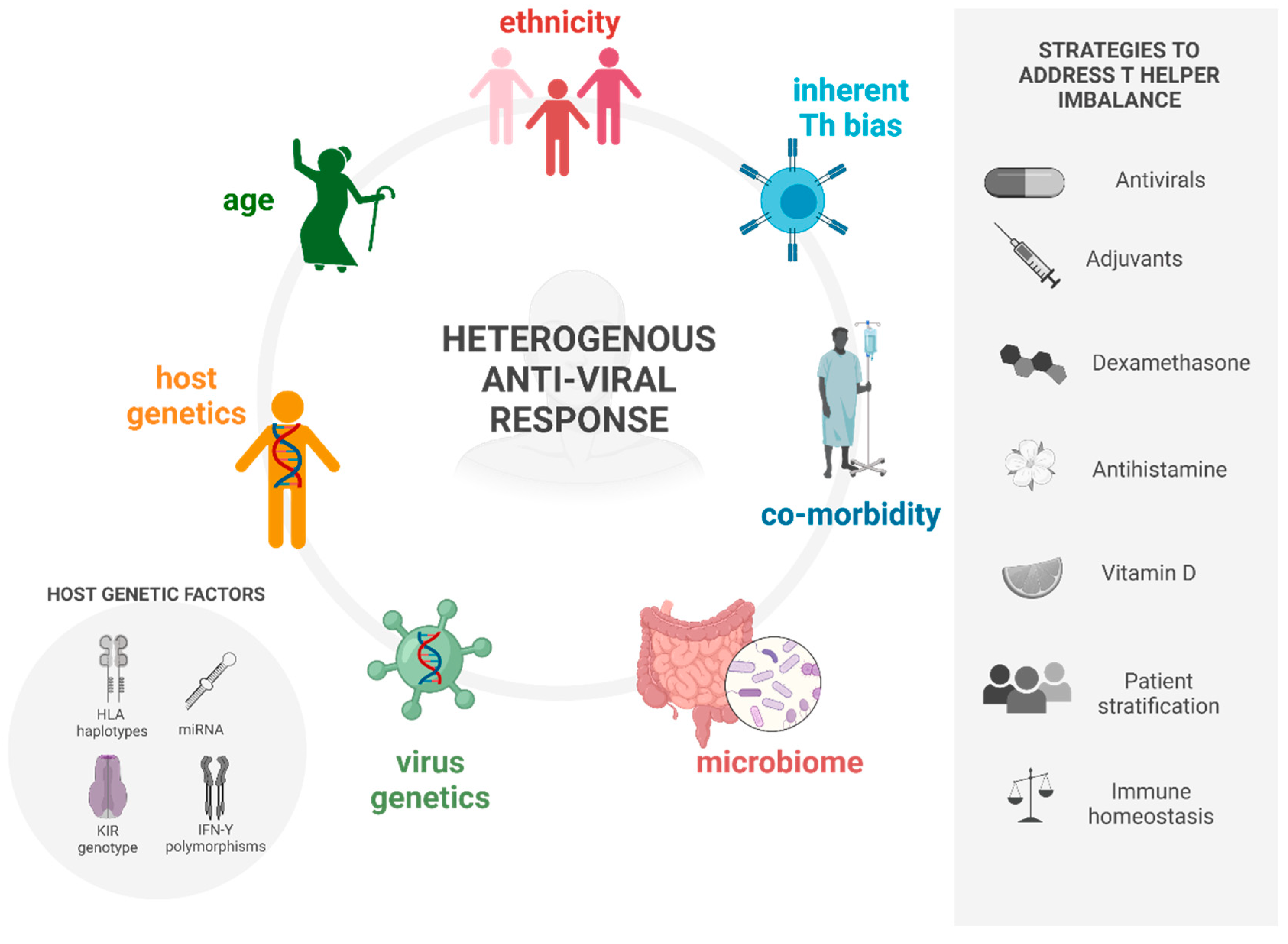
2. Causes of Deviant Immune Response in Viral Infections
2.1. Heterosubtypic Immunity
2.2. Ageing
2.3. Ethnicity
2.4. Co-Morbidities
3. Strategies to Address the Deviant Response
3.1. Antivirals
3.2. Anti-Histamines
3.3. Adjuvants
3.4. Vitamin D
3.5. Dexamethasone
3.6. Patient Stratification
4. Discussion and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Medzhitov, R.; Janeway, C.A., Jr. Innate immunity: Impact on the adaptive immune response. Curr. Opin. Immunol. 1997, 9, 4–9. [Google Scholar] [CrossRef]
- Swann, J.B.; Hayakawa, Y.; Zerafa, N.; Sheehan, K.C.; Scott, B.; Schreiber, R.D.; Hertzog, P.; Smyth, M.J. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J. Immunol. 2007, 178, 7540–7549. [Google Scholar] [CrossRef] [PubMed]
- Luft, T.; Pang, K.C.; Thomas, E.; Hertzog, P.; Hart, D.N.; Trapani, J.; Cebon, J. Type I IFNs enhance the terminal differentiation of dendritic cells. J. Immunol. 1998, 161, 1947–1953. [Google Scholar] [PubMed]
- Iezzi, G.; Karjalainen, K.; Lanzavecchia, A. The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity 1998, 8, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Thornley, T.B.; Phillips, N.E.; Beaudette-Zlatanova, B.C.; Markees, T.G.; Bahl, K.; Brehm, M.A.; Shultz, L.D.; Kurt-Jones, E.A.; Mordes, J.P.; Welsh, R.M.; et al. Type 1 IFN mediates cross-talk between innate and adaptive immunity that abrogates transplantation tolerance. J. Immunol. 2007, 179, 6620–6629. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Horvat, J.C.; Beagley, K.W.; Hansbro, P.M. Immunological decision-making: How does the immune system decide to mount a helper T-cell response? Immunology 2008, 123, 326–338. [Google Scholar] [CrossRef]
- McMichael, A.J.; Borrow, P.; Tomaras, G.D.; Goonetilleke, N.; Haynes, B.F. The immune response during acute HIV-1 infection: Clues for vaccine development. Nat. Rev. Immunol. 2010, 10, 11–23. [Google Scholar] [CrossRef]
- Rehermann, B.; Nascimbeni, M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 2005, 5, 215–229. [Google Scholar] [CrossRef]
- Hislop, A.D.; Taylor, G.S.; Sauce, D.; Rickinson, A.B. Cellular responses to viral infection in humans: Lessons from Epstein-Barr virus. Annu. Rev. Immunol. 2007, 25, 587–617. [Google Scholar] [CrossRef]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef]
- Kenney, A.D.; Dowdle, J.A.; Bozzacco, L.; McMichael, T.M.; Gelais, C.S.; Panfil, A.R.; Sun, Y.; Schlesinger, L.S.; Anderson, M.Z.; Green, P.L.; et al. Human Genetic Determinants of Viral Diseases. Annu. Rev. Genet. 2017, 51, 241–263. [Google Scholar] [CrossRef]
- Singh, R.; Kaul, R.; Kaul, A.; Khan, K. A comparative review of HLA associations with hepatitis B and C viral infections across global populations. World J. Gastroenterol. 2007, 13, 1770–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wit, J.; Borghans, J.A.; Kesmir, C.; van Baarle, D. Editorial: Role of HLA and KIR in Viral Infections. Front. Immunol. 2016, 7, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.H.; Zheng, X.; Ke, X.; Dorak, M.T.; Shen, J.; Boodram, B.; O’Gorman, M.; Beaman, K.; Cotler, S.J.; Hershow, R.; et al. Ethnic and geographical differences in HLA associations with the outcome of hepatitis C virus infection. Virol. J. 2009, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Catamo, E.; Zupin, L.; Freato, N.; Polesello, V.; Celsi, F.; Crocè, S.L.; Masutti, F.; Pozzato, G.; Segat, L.; Crovella, S. HLA-G regulatory polymorphisms are associated with susceptibility to HCV infection. HLA 2017, 89, 135–142. [Google Scholar] [CrossRef]
- Fernando, A.N.; Malavige, G.N.; Perera, K.L.; Premawansa, S.; Ogg, G.S.; De Silva, A.D. Polymorphisms of Transporter Associated with Antigen Presentation, Tumor Necrosis Factor-α and Interleukin-10 and their Implications for Protection and Susceptibility to Severe Forms of Dengue Fever in Patients in Sri Lanka. J. Glob. Infect. Dis. 2015, 7, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.C.; Huang, C.S.; Lee, C.N.; Chang, S.Y.; King, C.C.; Kao, C.L. Higher infection of dengue virus serotype 2 in human monocytes of patients with G6PD deficiency. PLoS ONE 2008, 3, e1557. [Google Scholar] [CrossRef]
- Wu, Y.H.; Tseng, C.P.; Cheng, M.L.; Ho, H.Y.; Shih, S.R.; Chiu, D.T. Glucose-6-phosphate dehydrogenase deficiency enhances human coronavirus 229E infection. J. Infect. Dis. 2008, 197, 812–816. [Google Scholar] [CrossRef] [Green Version]
- Ho, H.Y.; Cheng, M.L.; Weng, S.F.; Chang, L.; Yeh, T.T.; Shih, S.R.; Chiu, D.T. Glucose-6-phosphate dehydrogenase deficiency enhances enterovirus 71 infection. J. Gen. Virol. 2008, 89, 2080–2089. [Google Scholar] [CrossRef]
- Ávila-Pérez, G.; Nogales, A.; Park, J.-G.; Márquez-Jurado, S.; Iborra, F.J.; Almazan, F.; Martínez-Sobrido, L. A natural polymorphism in Zika virus NS2A protein responsible of virulence in mice. Sci. Rep. 2019, 9, 19968. [Google Scholar] [CrossRef]
- Alric, L.; Fort, M.; Izopet, J.; Vinel, J.P.; Charlet, J.P.; Selves, J.; Puel, J.; Pascal, J.P.; Duffaut, M.; Abbal, M. Genes of the major histocompatibility complex class II influence the outcome of hepatitis C virus infection. Gastroenterology 1997, 113, 1675–1681. [Google Scholar] [CrossRef]
- Kuzushita, N.; Hayashi, N.; Moribe, T.; Katayama, K.; Kanto, T.; Nakatani, S.; Kaneshige, T.; Tatsumi, T.; Ito, A.; Mochizuki, K.; et al. Influence of HLA haplotypes on the clinical courses of individuals infected with hepatitis C virus. Hepatology 1998, 27, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Asgari, S.; Schlapbach, L.J.; Anchisi, S.; Hammer, C.; Bartha, I.; Junier, T.; Mottet-Osman, G.; Posfay-Barbe, K.M.; Longchamp, D.; Stocker, M.; et al. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc. Natl. Acad. Sci. USA 2017, 114, 8342–8347. [Google Scholar] [CrossRef] [Green Version]
- Ciancanelli, M.J.; Huang, S.X.; Luthra, P.; Garner, H.; Itan, Y.; Volpi, S.; Lafaille, F.G.; Trouillet, C.; Schmolke, M.; Albrecht, R.A.; et al. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 2015, 348, 448–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, R.R. Age-related alterations in immune responses to West Nile virus infection. Clin. Exp. Immunol. 2017, 187, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brien, J.D.; Uhrlaub, J.L.; Hirsch, A.; Wiley, C.A.; Nikolich-Žugich, J. Key role of T cell defects in age-related vulnerability to West Nile virus. J. Exp. Med. 2009, 206, 2735–2745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, S.; Agrawal, S.; Cao, J.N.; Gupta, S.; Agrawal, A. Impaired secretion of interferons by dendritic cells from aged subjects to influenza: Role of histone modifications. Age 2013, 35, 1785–1797. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Jadhav, R.R.; Gustafson, C.E.; Smithey, M.J.; Hirsch, A.J.; Uhrlaub, J.L.; Hildebrand, W.H.; Nikolich-Žugich, J.; Weyand, C.M.; Goronzy, J.J. Defects in Antiviral T Cell Responses Inflicted by Aging-Associated miR-181a Deficiency. Cell Rep. 2019, 29, 2202–2216.e5. [Google Scholar] [CrossRef] [Green Version]
- Layden-Almer, J.E.; Ribeiro, R.M.; Wiley, T.; Perelson, A.S.; Layden, T.J. Viral dynamics and response differences in HCV-infected African American and white patients treated with IFN and ribavirin. Hepatology 2003, 37, 1343–1350. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 202, 756–759. [Google Scholar] [CrossRef]
- Haralambieva, I.H.; Salk, H.M.; Lambert, N.D.; Ovsyannikova, I.G.; Kennedy, R.B.; Warner, N.D.; Pankratz, V.S.; Poland, G.A. Associations between race, sex and immune response variations to rubella vaccination in two independent cohorts. Vaccine 2014, 32, 1946–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheridan, P.A.; Paich, H.A.; Handy, J.; Karlsson, E.A.; Hudgens, M.G.; Sammon, A.B.; Holland, L.A.; Weir, S.; Noah, T.L.; Beck, M.A. Obesity is associated with impaired immune response to influenza vaccination in humans. Int. J. Obes. 2012, 36, 1072–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuya, Y.; Roberts, S.; Hurteau, G.J.; Sanfilippo, A.M.; Racine, R.; Metzger, D.W. Asthma increases susceptibility to heterologous but not homologous secondary influenza. J. Virol. 2014, 88, 9166–9181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsumi, T.; Kierstead, L.S.; Ranieri, E.; Gesualdo, L.; Schena, F.P.; Finke, J.H.; Bukowski, R.M.; Mueller-Berghaus, J.; Kirkwood, J.M.; Kwok, W.W.; et al. Disease-associated bias in T helper type 1 (Th1)/Th2 CD4+ T cell responses against MAGE-6 in HLA-DRB10401+ patients with renal cell carcinoma or melanoma. J. Exp. Med. 2002, 196, 619–628. [Google Scholar] [CrossRef]
- Lan, F.; Zhang, N.; Gevaert, E.; Zhang, L.; Bachert, C. Viruses and bacteria in Th2-biased allergic airway disease. Allergy 2016, 71, 1381–1392. [Google Scholar] [CrossRef] [Green Version]
- Schlosser, J.; Dähnert, L.; Dremsek, P.; Tauscher, K.; Fast, C.; Ziegler, U.; Gröner, A.; Ulrich, R.G.; Groschup, M.H.; Eiden, M. Different Outcomes of Experimental Hepatitis E Virus Infection in Diverse Mouse Strains, Wistar Rats, and Rabbits. Viruses 2018, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Kulcsar, K.A.; Baxter, V.K.; Abraham, R.; Nelson, A.; Griffin, D.E. Distinct Immune Responses in Resistant and Susceptible Strains of Mice during Neurovirulent Alphavirus Encephalomyelitis. J. Virol. 2015, 89, 8280–8291. [Google Scholar] [CrossRef] [Green Version]
- Price, A.; Okumura, A.; Haddock, E.; Feldmann, F.; Meade-White, K.; Sharma, P.; Artami, M.; Lipkin, W.I.; Threadgill, D.W.; Feldmann, H.; et al. Transcriptional Correlates of Tolerance and Lethality in Mice Predict Ebola Virus Disease Patient Outcomes. Cell Rep. 2020, 30, 1702–1713.e6. [Google Scholar] [CrossRef] [Green Version]
- Collin, R.; Balmer, L.; Morahan, G.; Lesage, S. Common Heritable Immunological Variations Revealed in Genetically Diverse Inbred Mouse Strains of the Collaborative Cross. J. Immunol. 2019, 202, 777–786. [Google Scholar] [CrossRef] [Green Version]
- Olson, N.C.; Sallam, R.; Doyle, M.F.; Tracy, R.P.; Huber, S.A. T helper cell polarization in healthy people: Implications for cardiovascular disease. J. Cardiovasc. Transl. Res. 2013, 6, 772–786. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Kim, W.U. Microbiota in T-cell homeostasis and inflammatory diseases. Exp. Mol. Med. 2017, 49, e340. [Google Scholar] [CrossRef]
- Wilkinson, T.M.; Li, C.K.; Chui, C.S.; Huang, A.K.; Perkins, M.; Liebner, J.C.; Lambkin-Williams, R.; Gilbert, A.; Oxford, J.; Nicholas, B.; et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat. Med. 2012, 18, 274–280. [Google Scholar] [CrossRef]
- Epstein, S.L.; Lo, C.Y.; Misplon, J.A.; Lawson, C.M.; Hendrickson, B.A.; Max, E.E.; Subbarao, K. Mechanisms of heterosubtypic immunity to lethal influenza A virus infection in fully immunocompetent, T cell-depleted, beta2-microglobulin-deficient, and J chain-deficient mice. J. Immunol. 1997, 158, 1222–1230. [Google Scholar] [PubMed]
- Nguyen, H.H.; Moldoveanu, Z.; Novak, M.J.; van Ginkel, F.W.; Ban, E.; Kiyono, H.; McGhee, J.R.; Mestecky, J. Heterosubtypic immunity to lethal influenza A virus infection is associated with virus-specific CD8+ cytotoxic T lymphocyte responses induced in mucosa-associated tissues. Virology 1999, 254, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Borbet, T.C.; Fallegger, A.; Wipperman, M.F.; Blaser, M.J.; Müller, A. An Antibiotic-Impacted Microbiota Compromises the Development of Colonic Regulatory T Cells and Predisposes to Dysregulated Immune Responses. MBio 2021, 12, e03335-20. [Google Scholar] [CrossRef] [PubMed]
- Grayson, M.H.; Camarda, L.E.; Hussain, S.A.; Zemple, S.J.; Hayward, M.; Lam, V.; Hunter, D.A.; Santoro, J.L.; Rohlfing, M.; Cheung, D.S.; et al. Intestinal Microbiota Disruption Reduces Regulatory T Cells and Increases Respiratory Viral Infection Mortality Through Increased IFNγ Production. Front. Immunol. 2018, 9, 1587. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishna, C.; Kujawski, M.; Chu, H.; Li, L.; Mazmanian, S.K.; Cantin, E.M. Bacteroides fragilis polysaccharide A induces IL-10 secreting B and T cells that prevent viral encephalitis. Nat. Commun. 2019, 10, 2153. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, J.K.; Virgin, H.W. Viral immunity. Transkingdom control of viral infection and immunity in the mammalian intestine. Science 2016, 351, aad5872. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.; Diamond, M.S.; Thackray, L.B. Helminth-virus interactions: Determinants of coinfection outcomes. Gut Microbes 2021, 13, 1961202. [Google Scholar] [CrossRef]
- Drijvers, J.M.; Sharpe, A.H.; Haigis, M.C. The effects of age and systemic metabolism on anti-tumor T cell responses. Elife 2020, 9, e62420. [Google Scholar] [CrossRef]
- Hong, H.; Wang, Q.; Li, J.; Liu, H.; Meng, X.; Zhang, H. Aging, Cancer and Immunity. J. Cancer 2019, 10, 3021–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salam, N.; Rane, S.; Das, R.; Faulkner, M.; Gund, R.; Kandpal, U.; Lewis, V.; Mattoo, H.; Prabhu, S.; Ranganathan, V.; et al. T cell ageing: Effects of age on development, survival & function. Indian J. Med. Res. 2013, 138, 595–608. [Google Scholar] [PubMed]
- Tsukamoto, H.; Clise-Dwyer, K.; Huston, G.E.; Duso, D.K.; Buck, A.L.; Johnson, L.L.; Haynes, L.; Swain, S.L. Age-associated increase in lifespan of naïve CD4 T cells contributes to T-cell homeostasis but facilitates development of functional defects. Proc. Natl. Acad. Sci. USA 2009, 106, 18333–18338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, L.; Linton, P.J.; Eaton, S.M.; Tonkonogy, S.L.; Swain, S.L. Interleukin 2, but not other common γ chain-binding cytokines, can reverse the defect in generation of CD4 effector T cells from naive T cells of aged mice. J. Exp. Med. 1999, 190, 1013–1024. [Google Scholar] [CrossRef]
- Alberti, S.; Cevenini, E.; Ostan, R.; Capri, M.; Salvioli, S.; Bucci, L.; Ginaldi, L.; De Martinis, M.; Franceschi, C.; Monti, D. Age-dependent modifications of Type 1 and Type 2 cytokines within virgin and memory CD4+ T cells in humans. Mech. Ageing Dev. 2006, 127, 560–566. [Google Scholar] [CrossRef]
- Mansfield, A.S.; Nevala, W.K.; Dronca, R.S.; Leontovich, A.A.; Shuster, L.; Markovic, S.N. Normal ageing is associated with an increase in Th2 cells, MCP-1 (CCL1) and RANTES (CCL5), with differences in sCD40L and PDGF-AA between sexes. Clin. Exp. Immunol. 2012, 170, 186–193. [Google Scholar] [CrossRef]
- Eaton, S.M.; Burns, E.M.; Kusser, K.; Randall, T.D.; Haynes, L. Age-related defects in CD4 T cell cognate helper function lead to reductions in humoral responses. J. Exp. Med. 2004, 200, 1613–1622. [Google Scholar] [CrossRef] [Green Version]
- Müller, L.; Andrée, M.; Moskorz, W.; Drexler, I.; Walotka, L.; Grothmann, R.; Ptok, J.; Hillebrandt, J.; Ritchie, A.; Rabl, D.; et al. Age-dependent immune response to the Biontech/Pfizer BNT162b2 coronavirus disease 2019 vaccination. Clin. Infect. Dis. 2021, 73, 2065–2072. [Google Scholar] [CrossRef]
- Jergović, M.; Uhrlaub, J.L.; Watanabe, M.; Bradshaw, C.M.; White, L.M.; LaFleur, B.J.; Edwards, T.; Sprissler, R.; Worobey, M.; Bhattacharya, D.; et al. Competent immune responses to SARS-CoV-2 variants in older adults following two doses of mRNA vaccination. Nat. Commun. 2022, 13, 2891. [Google Scholar] [CrossRef]
- Chlibek, R.; Bayas, J.M.; Collins, H.; de la Pinta, M.L.; Ledent, E.; Mols, J.F.; Heineman, T.C. Safety and immunogenicity of an AS01-adjuvanted varicella-zoster virus subunit candidate vaccine against herpes zoster in adults ≥50 years of age. J. Infect. Dis. 2013, 208, 1953–1961. [Google Scholar] [CrossRef] [Green Version]
- Weinberger, B. Vaccines for the elderly: Current use and future challenges. Immun. Ageing 2018, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.L.; Linterman, M.A. Mechanisms underpinning poor antibody responses to vaccines in ageing. Immunol. Lett. 2022, 241, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sage, P.T.; Tan, C.L.; Freeman, G.J.; Haigis, M.; Sharpe, A.H. Defective TFH Cell Function and Increased TFR Cells Contribute to Defective Antibody Production in Aging. Cell Rep. 2015, 12, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, J.S.; Maue, A.C.; Eaton, S.M.; Lanthier, P.A.; Tighe, M.; Haynes, L. The aged microenvironment contributes to the age-related functional defects of CD4 T cells in mice. Aging Cell 2012, 11, 732–740. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, J.S.; Masters, A.R.; Hopkins, J.W.; Haynes, L. Age-related impairment of humoral response to influenza is associated with changes in antigen specific T follicular helper cell responses. Sci. Rep. 2016, 6, 25051. [Google Scholar] [CrossRef] [Green Version]
- Aydar, Y.; Balogh, P.; Tew, J.G.; Szakal, A.K. Altered regulation of Fc γ RII on aged follicular dendritic cells correlates with immunoreceptor tyrosine-based inhibition motif signaling in B cells and reduced germinal center formation. J. Immunol. 2003, 171, 5975–5987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stebegg, M.; Bignon, A.; Hill, D.L.; Silva-Cayetano, A.; Krueger, C.; Vanderleyden, I.; Innocentin, S.; Boon, L.; Wang, J.; Zand, M.S.; et al. Rejuvenating conventional dendritic cells and T follicular helper cell formation after vaccination. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Sakata-Kaneko, S.; Wakatsuki, Y.; Matsunaga, Y.; Usui, T.; Kita, T. Altered Th1/Th2 commitment in human CD4+ T cells with ageing. Clin. Exp. Immunol. 2000, 120, 267–273. [Google Scholar] [CrossRef]
- Baidya, S.G.; Zeng, Q.-T. Helper T cells and atherosclerosis: The cytokine web. Postgrad. Med. J. 2005, 81, 746–752. [Google Scholar] [CrossRef]
- Schulte, S.; Sukhova, G.K.; Libby, P. Genetically programmed biases in Th1 and Th2 immune responses modulate atherogenesis. Am. J. Pathol. 2008, 172, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Currier, N.L.; Sun, L.Z.; Miller, S.C. Exogenous melatonin: Quantitative enhancement in vivo of cells mediating non-specific immunity. J. Neuroimmunol. 2000, 104, 101–108. [Google Scholar] [CrossRef]
- Srinivasan, V.; Maestroni, G.J.; Cardinali, D.P.; Esquifino, A.I.; Perumal, S.R.; Miller, S.C. Melatonin, immune function and aging. Immun. Ageing 2005, 2, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.-H.; Liao, C.-L.; Chen, S.-J.; Shi, L.-G.; Lin, L.; Chen, Y.-W.; Cheng, C.-P.; Sytwu, H.-K.; Shang, S.-T.; Lin, G.-J. Melatonin possesses an anti-influenza potential through its immune modulatory effect. J. Funct. Foods 2019, 58, 189–198. [Google Scholar] [CrossRef]
- Luo, P.; Qiu, L.; Liu, Y.; Liu, X.L.; Zheng, J.L.; Xue, H.Y.; Liu, W.H.; Liu, D.; Li, J. Metformin Treatment Was Associated with Decreased Mortality in COVID-19 Patients with Diabetes in a Retrospective Analysis. Am. J. Trop. Med. Hyg. 2020, 103, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Bramante, C.T.; Ingraham, N.E.; Murray, T.A.; Marmor, S.; Hovertsen, S.; Gronski, J.; McNeil, C.; Feng, R.; Guzman, G.; Abdelwahab, N.; et al. Observational Study of Metformin and Risk of Mortality in Patients Hospitalized with Covid-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Zhou, X.; McElhaney, J.E. Age-related changes in memory and effector T cells responding to influenza A/H3N2 and pandemic A/H1N1 strains in humans. Vaccine 2011, 29, 2169–2177. [Google Scholar] [CrossRef] [Green Version]
- Helfand, B.K.I.; Webb, M.; Gartaganis, S.L.; Fuller, L.; Kwon, C.S.; Inouye, S.K. The Exclusion of Older Persons From Vaccine and Treatment Trials for Coronavirus Disease 2019—Missing the Target. JAMA Intern. Med. 2020, 180, 1546–1549. [Google Scholar] [CrossRef]
- Bhopal, R. Glossary of terms relating to ethnicity and race: For reflection and debate. J. Epidemiol. Community Health 2004, 58, 441–445. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Rotundo, L.; Yang, J.D.; Kim, D.; Kothari, N.; Feurdean, M.; Ruhl, C.; Unalp-Arida, A. Racial/ethnic disparities in the prevalence and awareness of Hepatitis B virus infection and immunity in the United States. J. Viral Hepat. 2017, 24, 1052–1066. [Google Scholar] [CrossRef]
- Kawsar, M.; Goh, B. Hepatitis B virus infection among Chinese residents in the United Kingdom. Sex. Transm. Infect. 2002, 78, 166–168. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.T.; Loggi, E.; Boni, C.; Chia, A.; Gehring, A.J.; Sastry, K.S.; Goh, V.; Fisicaro, P.; Andreone, P.; Brander, C.; et al. Host ethnicity and virus genotype shape the hepatitis B virus-specific T-cell repertoire. J. Virol. 2008, 82, 10986–10997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, M.; Wood, R.; Myer, L.; Taffé, P.; Rauch, A.; Battegay, M.; Egger, M.; Cape Town AIDS Cohort and the Swiss HIV Cohort Study. CD4+ T cell count decreases by ethnicity among untreated patients with HIV infection in South Africa and Switzerland. J. Infect. Dis. 2009, 200, 1729–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, C.; Casado, J.L.; Härter, G.; Vizcarra, P.; Moreno, A.; Cattaneo, D.; Meraviglia, P.; Spinner, C.D.; Schabaz, F.; Grunwald, S.; et al. Immune deficiency is a risk factor for severe COVID-19 in people living with HIV. HIV Med. 2021, 22, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Rydyznski Moderbacher, C.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J.; et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 2020, 183, 996–1012.e19. [Google Scholar] [CrossRef] [PubMed]
- Fendler, A.; Shepherd, S.T.C.; Au, L.; Wilkinson, K.A.; Wu, M.; Byrne, F.; Cerrone, M.; Schmitt, A.M.; Joharatnam-Hogan, N.; Shum, B.; et al. Adaptive immunity and neutralizing antibodies against SARS-CoV-2 variants of concern following vaccination in patients with cancer: The CAPTURE study. Nat. Cancer 2021, 2, 1305–1320. [Google Scholar] [CrossRef] [PubMed]
- Calvet, J.; Gratacós, J.; Amengual, M.J.; Llop, M.; Navarro, M.; Moreno, A.; Berenguer-Llergo, A.; Serrano, A.; Orellana, C.; Cervantes, M. CD4 and CD8 Lymphocyte Counts as Surrogate Early Markers for Progression in SARS-CoV-2 Pneumonia: A Prospective Study. Viruses 2020, 12, 1277. [Google Scholar] [CrossRef] [PubMed]
- Sze, S.; Pan, D.; Nevill, C.R.; Gray, L.J.; Martin, C.A.; Nazareth, J.; Minhas, J.S.; Divall, P.; Khunti, K.; Abrams, K.R.; et al. Ethnicity and clinical outcomes in COVID-19: A systematic review and meta-analysis. EClinicalMedicine 2020, 29, 100630. [Google Scholar] [CrossRef]
- Shrotri, M.; van Schalkwyk, M.C.I.; Post, N.; Eddy, D.; Huntley, C.; Leeman, D.; Rigby, S.; Williams, S.V.; Bermingham, W.H.; Kellam, P.; et al. T cell response to SARS-CoV-2 infection in humans: A systematic review. PLoS ONE 2021, 16, e0245532. [Google Scholar] [CrossRef]
- Park, S.Y.; Boushey, C.J.; Shvetsov, Y.B.; Wirth, M.D.; Shivappa, N.; Hébert, J.R.; Haiman, C.A.; Wilkens, L.R.; Le Marchand, L. Diet Quality and Risk of Lung Cancer in the Multiethnic Cohort Study. Nutrients 2021, 13, 1614. [Google Scholar] [CrossRef]
- Ojo, O. Diabetes in Ethnic Minorities in UK: The Role of Diet in Glucose Dysregulation and Prevalence of Diabetes. J. Food Nutr. Disord. 2013, 2, 2. [Google Scholar] [CrossRef]
- Chaturvedi, N. Ethnic differences in cardiovascular disease. Heart 2003, 89, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kang, M.; Wilkens, L.R.; Shvetsov, Y.B.; Harmon, B.E.; Shivappa, N.; Wirth, M.D.; Hébert, J.R.; Haiman, C.A.; Le Marchand, L.; et al. The Dietary Inflammatory Index and All-Cause, Cardiovascular Disease, and Cancer Mortality in the Multiethnic Cohort Study. Nutrients 2018, 10, 1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Montero, C.; Fraile-Martínez, O.; Gómez-Lahoz, A.; Pekarek, L.; Castellanos, A.; Noguerales-Fraguas, F.; Coca, S.; Guijarro, L.; García-Honduvilla, N.; Asúnsolo, A.; et al. Nutritional Components in Western Diet Versus Mediterranean Diet at the Gut Microbiota-Immune System Interplay. Implications for Health and Disease. Nutrients 2021, 13, 699. [Google Scholar] [CrossRef] [PubMed]
- Piyathilake, C.J.; Badiga, S.; Chappell, A.R.; Johanning, G.L.; Jolly, P.E. Racial differences in dietary choices and their relationship to inflammatory potential in childbearing age women at risk for exposure to COVID-19. Nutr. Res. 2021, 90, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Arroyo Hornero, R.; Hamad, I.; Côrte-Real, B.; Kleinewietfeld, M. The Impact of Dietary Components on Regulatory T Cells and Disease. Front. Immunol. 2020, 11, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, P.M.; Guttman-Yassky, E. Racial differences in atopic dermatitis. Ann. Allergy Asthma Immunol. 2019, 122, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, R.D.; Pavel, A.B.; Glickman, J.; Chan, T.C.; Zheng, X.; Zhang, N.; Cueto, I.; Peng, X.; Estrada, Y.; Fuentes-Duculan, J.; et al. Atopic dermatitis in African American patients is TH2/TH22-skewed with TH1/TH17 attenuation. Ann. Allergy Asthma Immunol. 2019, 122, 99–110.e6. [Google Scholar] [CrossRef] [Green Version]
- Meditz, A.L.; MaWhinney, S.; Allshouse, A.; Feser, W.; Markowitz, M.; Little, S.; Connick, E. Sex, race, and geographic region influence clinical outcomes following primary HIV-1 infection. J. Infect. Dis. 2011, 203, 442–451. [Google Scholar] [CrossRef]
- Mathew, A.; Townsley, E.; Ennis, F.A. Elucidating the role of T cells in protection against and pathogenesis of dengue virus infections. Future Microbiol. 2014, 9, 411–425. [Google Scholar] [CrossRef] [Green Version]
- Sierra, B.D.; Kouri, G.; Guzmán, M.G. Race: A risk factor for dengue hemorrhagic fever. Arch. Virol. 2007, 152, 533–542. [Google Scholar] [CrossRef]
- Erener, S. Diabetes, infection risk and COVID-19. Mol. Metab. 2020, 39, 101044. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Mahawar, K.; Xia, Z.; Yang, W.; El-Hasani, S. RETRACTED: Obesity and mortality of COVID-19. Meta-analysis. Obes. Res. Clin. Pract. 2020, 14, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.L. Asthma and COVID-19: Is asthma a risk factor for severe outcomes? Allergy 2020, 75, 1543–1545. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.A.; Dickinson, A.K.; Taams, L.S. The Interplay Between Monocytes/Macrophages and CD4+ T Cell Subsets in Rheumatoid Arthritis. Front. Immunol. 2015, 6, 571. [Google Scholar] [CrossRef] [PubMed]
- Savinov, A.Y.; Wong, F.S.; Chervonsky, A.V. IFN-γ affects homing of diabetogenic T cells. J. Immunol. 2001, 167, 6637–6643. [Google Scholar] [CrossRef] [Green Version]
- Kunkl, M.; Frascolla, S.; Amormino, C.; Volpe, E.; Tuosto, L. T Helper Cells: The Modulators of Inflammation in Multiple Sclerosis. Cells 2020, 9, 482. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Luo, Y.; Xu, H.; Ma, Q.; Yao, Q. Imbalance between T helper 1 and regulatory T cells plays a detrimental role in experimental Parkinson’s disease in mice. J. Int. Med. Res. 2021, 49, 300060521998471. [Google Scholar] [CrossRef]
- Williams, G.P.; Schonhoff, A.M.; Jurkuvenaite, A.; Gallups, N.J.; Standaert, D.G.; Harms, A.S. CD4 T cells mediate brain inflammation and neurodegeneration in a mouse model of Parkinson’s disease. Brain 2021, 144, 2047–2059. [Google Scholar] [CrossRef]
- León, B.; Ballesteros-Tato, A. Modulating Th2 Cell Immunity for the Treatment of Asthma. Front. Immunol. 2021, 12, 637948. [Google Scholar] [CrossRef]
- Seumois, G.; Zapardiel-Gonzalo, J.; White, B.; Singh, D.; Schulten, V.; Dillon, M.; Hinz, D.; Broide, D.H.; Sette, A.; Peters, B.; et al. Transcriptional Profiling of Th2 Cells Identifies Pathogenic Features Associated with Asthma. J. Immunol. 2016, 197, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Li, M.G.; Liu, X.Y.; Liu, Z.Q.; Hong, J.Y.; Liu, J.Q.; Zhou, C.J.; Hu, T.Y.; Xiao, X.J.; Ran, P.X.; Zheng, P.Y.; et al. Bcl2L12 Contributes to Th2-Biased Inflammation in the Intestinal Mucosa by Regulating CD4+ T Cell Activities. J. Immunol. 2018, 201, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Harding, S.; Sorenson, M.; Jason, L.A.; Maher, K.; Fletcher, M.A. Evidence for T-helper 2 shift and association with illness parameters in chronic fatigue syndrome (CFS). Bull. IACFS/ME 2008, 16, 19–33. [Google Scholar]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: Cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naïve and drug-treated patients. J. Neuroinflammation 2018, 15, 205. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, Z.; Wang, X.Q.; Qiu, Y.H.; Peng, Y.P. A dysfunction of CD4+ T lymphocytes in peripheral immune system of Parkinson’s disease model mice. Chin. J. Appl. Physiol. 2014, 30, 567–576. [Google Scholar]
- Huber, S.A.; Sakkinen, P.; David, C.; Newell, M.K.; Tracy, R.P. T helper-cell phenotype regulates atherosclerosis in mice under conditions of mild hypercholesterolemia. Circulation 2001, 103, 2610–2616. [Google Scholar] [CrossRef] [Green Version]
- Buono, C.; Binder, C.J.; Stavrakis, G.; Witztum, J.L.; Glimcher, L.H.; Lichtman, A.H. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc. Natl. Acad. Sci. USA 2005, 102, 1596–1601. [Google Scholar] [CrossRef] [Green Version]
- Soejima, H.; Irie, A.; Miyamoto, S.; Kajiwara, I.; Kojima, S.; Hokamaki, J.; Sakamoto, T.; Tanaka, T.; Yoshimura, M.; Nishimura, Y.; et al. Preference toward a T-helper type 1 response in patients with coronary spastic angina. Circulation 2003, 107, 2196–2200. [Google Scholar] [CrossRef]
- Methe, H.; Brunner, S.; Wiegand, D.; Nabauer, M.; Koglin, J.; Edelman, E.R. Enhanced T-helper-1 lymphocyte activation patterns in acute coronary syndromes. J. Am. Coll. Cardiol. 2005, 45, 1939–1945. [Google Scholar] [CrossRef] [Green Version]
- Benagiano, M.; Azzurri, A.; Ciervo, A.; Amedei, A.; Tamburini, C.; Ferrari, M.; Telford, J.L.; Baldari, C.T.; Romagnani, S.; Cassone, A.; et al. T helper type 1 lymphocytes drive inflammation in human atherosclerotic lesions. Proc. Natl. Acad. Sci. USA 2003, 100, 6658–6663. [Google Scholar] [CrossRef] [Green Version]
- Engelbertsen, D.; Andersson, L.; Ljungcrantz, I.; Wigren, M.; Hedblad, B.; Nilsson, J.; Björkbacka, H. T-helper 2 immunity is associated with reduced risk of myocardial infarction and stroke. Arter. Thromb. Vasc. Biol. 2013, 33, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Pène, J.; Chevalier, S.; Preisser, L.; Vénéreau, E.; Guilleux, M.-H.; Ghannam, S.; Molès, J.-P.; Danger, Y.; Ravon, E.; Lesaux, S.; et al. Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J. Immunol. 2008, 180, 7423–7430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, J.; Wang, K.; Meng, Q.H.; Qi, Z.X.; Meng, F.L.; Fan, Y.C. Implication of Th17 and Th1 cells in patients with chronic active hepatitis B. J. Clin. Immunol. 2010, 30, 60–67. [Google Scholar] [CrossRef]
- Wang, H.; Liu, X.; Tan, C.; Zhou, W.; Jiang, J.; Peng, W.; Zhou, X.; Mo, L.; Chen, L. Bacterial, viral, and fungal infection-related risk of Parkinson’s disease: Meta-analysis of cohort and case-control studies. Brain Behav. 2020, 10, e01549. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Mai, T.H.; Kim, K.; Cho, H.; Ki, M. Association between viral hepatitis infection and Parkinson’s disease: A population-based prospective study. J. Viral Hepat. 2020, 27, 1171–1178. [Google Scholar] [CrossRef]
- Tracy, R.P.; Doyle, M.F.; Olson, N.C.; Huber, S.A.; Jenny, N.S.; Sallam, R.; Psaty, B.M.; Kronmal, R.A. T-helper type 1 bias in healthy people is associated with cytomegalovirus serology and atherosclerosis: The Multi-Ethnic Study of Atherosclerosis. J. Am. Heart Assoc. 2013, 2, e000117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shurin, M.R.; Lu, L.; Kalinski, P.; Stewart-Akers, A.M.; Lotze, M.T. Th1/Th2 balance in cancer, transplantation and pregnancy. Springer Semin. Immunopathol. 1999, 21, 339–359. [Google Scholar] [CrossRef]
- Shimato, S.; Maier, L.M.; Maier, R.; Bruce, J.N.; Anderson, R.C.; Anderson, D.E. Profound tumor-specific Th2 bias in patients with malignant glioma. BMC Cancer 2012, 12, 561. [Google Scholar] [CrossRef] [Green Version]
- Nevala, W.K.; Vachon, C.M.; Leontovich, A.A.; Scott, C.G.; Thompson, M.A.; Markovic, S.N.; for the Melanoma Study Group of the Mayo Clinic Cancer Center. Evidence of systemic Th2-driven chronic inflammation in patients with metastatic melanoma. Clin. Cancer Res. 2009, 15, 1931–1939. [Google Scholar] [CrossRef] [Green Version]
- Guenova, E.; Watanabe, R.; Teague, J.E.; Desimone, J.A.; Jiang, Y.; Dowlatshahi, M.; Schlapbach, C.; Schaekel, K.; Rook, A.H.; Tawa, M.; et al. TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clin. Cancer Res. 2013, 19, 3755–3763. [Google Scholar] [CrossRef] [Green Version]
- Glajcar, A.; Szpor, J.; Hodorowicz-Zaniewska, D.; Tyrak, K.E.; Okoń, K. The composition of T cell infiltrates varies in primary invasive breast cancer of different molecular subtypes as well as according to tumor size and nodal status. Virchows Arch. 2019, 475, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Lathers, D.M.; Achille, N.J.; Young, M.R. Incomplete Th2 skewing of cytokines in plasma of patients with squamous cell carcinoma of the head and neck. Hum. Immunol. 2003, 64, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.C.; Yao, S.; McCann, S.E.; Dolnick, R.Y.; Wallace, P.K.; Gong, Z.; Quan, L.; Lee, K.P.; Evans, S.S.; Repasky, E.A.; et al. Pretreatment levels of circulating Th1 and Th2 cytokines, and their ratios, are associated with ER-negative and triple negative breast cancers. Breast Cancer Res. Treat. 2013, 139, 477–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snell, L.M.; Osokine, I.; Yamada, D.H.; De la Fuente, J.R.; Elsaesser, H.J.; Brooks, D.G. Overcoming CD4 Th1 Cell Fate Restrictions to Sustain Antiviral CD8 T Cells and Control Persistent Virus Infection. Cell Rep. 2016, 16, 3286–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.B.; Puzanov, I.; Kelley, M.C. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy 2015, 7, 611–619. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.N.; Gillespie, G.Y.; Love, C.E.; Randall, S.; Whitley, R.J.; Markert, J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 2208–2213. [Google Scholar] [CrossRef] [Green Version]
- Grødeland, G.; Fossum, E.; Bogen, B. Polarizing T and B Cell Responses by APC-Targeted Subunit Vaccines. Front. Immunol. 2015, 6, 367. [Google Scholar] [CrossRef]
- Shi, L.; Xiong, H.; He, J.; Deng, H.; Li, Q.; Zhong, Q.; Hou, W.; Cheng, L.; Xiao, H.; Yang, Z. Antiviral activity of arbidol against influenza A virus, respiratory syncytial virus, rhinovirus, coxsackie virus and adenovirus in vitro and in vivo. Arch. Virol. 2007, 152, 1447–1455. [Google Scholar] [CrossRef]
- Brooks, M.J.; Burtseva, E.I.; Ellery, P.J.; Marsh, G.A.; Lew, A.M.; Slepushkin, A.N.; Crowe, S.M.; Tannock, G.A. Antiviral activity of arbidol, a broad-spectrum drug for use against respiratory viruses, varies according to test conditions. J. Med. Virol. 2012, 84, 170–181. [Google Scholar] [CrossRef]
- Liu, Q.; Xiong, H.R.; Lu, L.; Liu, Y.Y.; Luo, F.; Hou, W.; Yang, Z.Q. Antiviral and anti-inflammatory activity of arbidol hydrochloride in influenza A (H1N1) virus infection. Acta Pharmacol. Sin. 2013, 34, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ding, Y.; Yang, C.; Li, R.; Du, Q.; Hao, Y.; Li, Z.; Jiang, H.; Zhao, J.; Chen, Q.; et al. Inhibition of the infectivity and inflammatory response of influenza virus by Arbidol hydrochloride in vitro and in vivo (mice and ferret). Biomed. Pharmacother. 2017, 91, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Lopalco, G.; Lucherini, O.M.; Lopalco, A.; Venerito, V.; Fabiani, C.; Frediani, B.; Galeazzi, M.; Lapadula, G.; Cantarini, L.; Iannone, F. Cytokine Signatures in Mucocutaneous and Ocular Behçet’s Disease. Front. Immunol. 2017, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.L.; Smith, D.W.; Cummins, M.J.; Jacoby, P.A.; Cummins, J.M.; Beilharz, M.W. Low-dose oral interferon alpha as prophylaxis against viral respiratory illness: A double-blind, parallel controlled trial during an influenza pandemic year. Influenza Other Respir. Viruses 2013, 7, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Touzot, M.; Cacoub, P.; Bodaghi, B.; Soumelis, V.; Saadoun, D. IFN-α, induces IL-10 production and tilt the balance between Th1 and Th17 in Behçet disease. Autoimmun. Rev. 2015, 14, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Kaushal, P.; Agrawal, S.; Gollapudi, S.; Gupta, S. Thimerosal induces TH2 responses via influencing cytokine secretion by human dendritic cells. J. Leukoc. Biol. 2007, 81, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, A.; Ferraz-De-Paula, V.; Pinheiro, M.L.; Vitoretti, L.B.; Mariano-Souza, D.P.; Quinteiro-Filho, W.M.; Akamine, A.T.; Almeida, V.I.; Quevedo, J.; Dal-Pizzol, F.; et al. Cannabidiol, a non-psychotropic plant-derived cannabinoid, decreases inflammation in a murine model of acute lung injury: Role for the adenosine A2A receptor. Eur. J. Pharmacol. 2012, 678, 78–85. [Google Scholar] [CrossRef]
- Yuan, M.; Kiertscher, S.M.; Cheng, Q.; Zoumalan, R.; Tashkin, D.P.; Roth, M.D. Δ9-Tetrahydrocannabinol regulates Th1/Th2 cytokine balance in activated human T cells. J. Neuroimmunol. 2002, 133, 124–131. [Google Scholar] [CrossRef]
- Tahamtan, A.; Tavakoli-Yaraki, M.; Rygiel, T.P.; Mokhtari-Azad, T.; Salimi, V. Effects of cannabinoids and their receptors on viral infections. J. Med. Virol. 2016, 88, 1–12. [Google Scholar] [CrossRef]
- He, S.; Lin, B.; Chu, V.; Hu, Z.; Hu, X.; Xiao, J.; Wang, A.Q.; Schweitzer, C.J.; Li, Q.; Imamura, M.; et al. Repurposing of the antihistamine chlorcyclizine and related compounds for treatment of hepatitis C virus infection. Sci. Transl. Med. 2015, 7, 282ra49. [Google Scholar] [CrossRef] [Green Version]
- Klemenstsson, H.; Andersson, M.; Pipkorn, U. Allergen-induced increase in nonspecific nasal reactivity is blocked by antihistamines without a clear-cut relationship to eosinophil influx. J. Allergy Clin. Immunol. 1990, 86, 466–472. [Google Scholar] [CrossRef]
- Robinson, D.; Hamid, Q.; Bentley, A.; Ying, S.; Kay, A.B.; Durham, S.R. Activation of CD4+ T cells, increased TH2-type cytokine mRNA expression, and eosinophil recruitment in bronchoalveolar lavage after allergen inhalation challenge in patients with atopic asthma. J. Allergy Clin. Immunol. 1993, 92, 313–324. [Google Scholar] [CrossRef]
- Abdelaziz, M.M.; Devalia, J.L.; Khair, O.A.; Bayram, H.; Prior, A.J.; Davies, R.J. Effect of fexofenadine on eosinophil-induced changes in epithelial permeability and cytokine release from nasal epithelial cells of patients with seasonal allergic rhinitis. J. Allergy Clin. Immunol. 1998, 101, 410–420. [Google Scholar] [CrossRef]
- Kato, Y.; Manabe, T.; Tanaka, Y.; Mochizuki, H. Effect of an orally active Th1/Th2 balance modulator, M50367, on IgE production, eosinophilia, and airway hyperresponsiveness in mice. J. Immunol. 1999, 162, 7470–7479. [Google Scholar] [PubMed]
- Xu, W.; Xia, S.; Pu, J.; Wang, Q.; Li, P.; Lu, L.; Jiang, S. The Antihistamine Drugs Carbinoxamine Maleate and Chlorpheniramine Maleate Exhibit Potent Antiviral Activity Against a Broad Spectrum of Influenza Viruses. Front. Microbiol. 2018, 9, 2643. [Google Scholar] [CrossRef] [PubMed]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine adjuvants: Putting innate immunity to work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Grun, J.L.; Maurer, P.H. Different T helper cell subsets elicited in mice utilizing two different adjuvant vehicles: The role of endogenous interleukin 1 in proliferative responses. Cell. Immunol. 1989, 121, 134–145. [Google Scholar] [CrossRef]
- Kool, M.; Soullié, T.; van Nimwegen, M.; Willart, M.A.; Muskens, F.; Jung, S.; Hoogsteden, H.C.; Hammad, H.; Lambrecht, B.N. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J. Exp. Med. 2008, 205, 869–882. [Google Scholar] [CrossRef] [Green Version]
- Sharp, F.A.; Ruane, D.; Claass, B.; Creagh, E.; Harris, J.; Malyala, P.; Singh, M.; O’Hagan, D.T.; Pétrilli, V.; Tschopp, J.; et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 870–875. [Google Scholar] [CrossRef] [Green Version]
- Jordan, M.B.; Mills, D.M.; Kappler, J.; Marrack, P.; Cambier, J.C. Promotion of B cell immune responses via an alum-induced myeloid cell population. Science 2004, 304, 1808–1810. [Google Scholar] [CrossRef]
- Didierlaurent, A.M.; Morel, S.; Lockman, L.; Giannini, S.L.; Bisteau, M.; Carlsen, H.; Kielland, A.; Vosters, O.; Vanderheyde, N.; Schiavetti, F.; et al. AS04, an aluminum salt- and TLR4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J. Immunol. 2009, 183, 6186–6197. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Van der Meer, J.W.; Sutmuller, R.P.; Adema, G.J.; Kullberg, B.J. From the Th1/Th2 paradigm towards a Toll-like receptor/T-helper bias. Antimicrob. Agents Chemother. 2005, 49, 3991–3996. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, I.; Garg, R.; van Drunen Littel-van den Hurk, S. Selection of adjuvants for vaccines targeting specific pathogens. Expert Rev. Vaccines 2019, 18, 505–521. [Google Scholar] [CrossRef] [PubMed]
- Janovec, V.; Hodek, J.; Clarova, K.; Hofman, T.; Dostalik, P.; Fronek, J.; Chlupac, J.; Chaperot, L.; Durand, S.; Baumert, T.F.; et al. Toll-like receptor dual-acting agonists are potent inducers of PBMC-produced cytokines that inhibit hepatitis B virus production in primary human hepatocytes. Sci. Rep. 2020, 10, 12767. [Google Scholar] [CrossRef] [PubMed]
- Ratnapriya, S.; Perez-Greene, E.; Schifanella, L.; Herschhorn, A. Adjuvant-mediated enhancement of the immune response to HIV vaccines. FEBS J. 2021, 289, 3317–3334. [Google Scholar] [CrossRef] [PubMed]
- Coler, R.N.; Baldwin, S.L.; Shaverdian, N.; Bertholet, S.; Reed, S.J.; Raman, V.S.; Lu, X.; DeVos, J.; Hancock, K.; Katz, J.M.; et al. A synthetic adjuvant to enhance and expand immune responses to influenza vaccines. PLoS ONE 2010, 5, e13677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapuy, M.C.; Arlot, M.E.; Duboeuf, F.; Brun, J.; Crouzet, B.; Arnaud, S.; Delmas, P.D.; Meunier, P.J. Vitamin D3 and calcium to prevent hip fractures in elderly women. N. Engl. J. Med. 1992, 327, 1637–1642. [Google Scholar] [CrossRef]
- Hansdottir, S.; Monick, M.M.; Hinde, S.L.; Lovan, N.; Look, D.C.; Hunninghake, G.W. Respiratory epithelial cells convert inactive vitamin D to its active form: Potential effects on host defense. J. Immunol. 2008, 181, 7090–7099. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef]
- Yim, S.; Dhawan, P.; Ragunath, C.; Christakos, S.; Diamond, G. Induction of cathelicidin in normal and CF bronchial epithelial cells by 1,25-dihydroxyvitamin D3. J. Cyst. Fibros. 2007, 6, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Hansdottir, S.; Monick, M.M.; Lovan, N.; Powers, L.; Gerke, A.; Hunninghake, G.W. Vitamin D decreases respiratory syncytial virus induction of NF-κB-linked chemokines and cytokines in airway epithelium while maintaining the antiviral state. J. Immunol. 2010, 184, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Rehman, P.K. Sub-clinical rickets and recurrent infection. J. Trop. Pediatr. 1994, 40, 58. [Google Scholar] [CrossRef]
- Laaksi, I.; Ruohola, J.P.; Tuohimaa, P.; Auvinen, A.; Haataja, R.; Pihlajamäki, H.; Ylikomi, T. An association of serum vitamin D concentrations <40 nmol/L with acute respiratory tract infection in young Finnish men. Am. J. Clin. Nutr. 2007, 86, 714–717. [Google Scholar] [CrossRef] [Green Version]
- Roth, D.E.; Shah, R.; Black, R.E.; Baqui, A.H. Vitamin D status and acute lower respiratory infection in early childhood in Sylhet, Bangladesh. Acta Paediatr. 2010, 99, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Wayse, V.; Yousafzai, A.; Mogale, K.; Filteau, S. Association of subclinical vitamin D deficiency with severe acute lower respiratory infection in Indian children under 5 y. Eur. J. Clin. Nutr. 2004, 58, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karatekin, G.; Kaya, A.; Salihoglu, O.; Balci, H.; Nuhoglu, A. Association of subclinical vitamin D deficiency in newborns with acute lower respiratory infection and their mothers. Eur. J. Clin. Nutr. 2009, 63, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Avenell, A.; Cook, J.A.; Maclennan, G.S.; Macpherson, G.C. Vitamin D supplementation to prevent infections: A sub-study of a randomised placebo-controlled trial in older people (RECORD trial, ISRCTN 51647438). Age Ageing 2007, 36, 574–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parva, N.R.; Tadepalli, S.; Singh, P.; Qian, A.; Joshi, R.; Kandala, H.; Nookala, V.K.; Cheriyath, P. Prevalence of Vitamin D Deficiency and Associated Risk Factors in the US Population (2011–2012). Cureus 2018, 10, e2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li-Ng, M.; Aloia, J.F.; Pollack, S.; Cunha, B.A.; Mikhail, M.; Yeh, J.; Berbari, N. A randomized controlled trial of vitamin D3 supplementation for the prevention of symptomatic upper respiratory tract infections. Epidemiol. Infect. 2009, 137, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, A.; Barrat, F.J.; Crain, C.; Heath, V.L.; Savelkoul, H.F.; O’Garra, A. 1α,25-Dihydroxyvitamin d3 has a direct effect on naive CD4+ T cells to enhance the development of Th2 cells. J. Immunol. 2001, 167, 4974–4980. [Google Scholar] [CrossRef] [Green Version]
- Zdrenghea, M.T.; Makrinioti, H.; Bagacean, C.; Bush, A.; Johnston, S.L.; Stanciu, L.A. Vitamin D modulation of innate immune responses to respiratory viral infections. Rev. Med. Virol. 2017, 27, e1909. [Google Scholar] [CrossRef]
- Teymoori-Rad, M.; Shokri, F.; Salimi, V.; Marashi, S.M. The interplay between vitamin D and viral infections. Rev. Med. Virol. 2019, 29, e2032. [Google Scholar] [CrossRef]
- Lee, C. Controversial Effects of Vitamin D and Related Genes on Viral Infections, Pathogenesis, and Treatment Outcomes. Nutrients 2020, 12, 962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Blotta, H.M.; Mamoni, R.L.; Louzada, P.; Bertolo, M.B.; Foss, N.T.; Moreira, A.C.; Castro, M. Effects of dexamethasone on lymphocyte proliferation and cytokine production in rheumatoid arthritis. J. Rheumatol. 2002, 29, 46–51. [Google Scholar]
- Nair, R.; Kakroo, A.; Bapna, A.; Gogia, A.; Vora, A.; Pathak, A.; Korula, A.; Chakrapani, A.; Doval, D.; Prakash, G.; et al. Management of Lymphomas: Consensus Document 2018 by an Indian Expert Group. Indian J. Hematol. Blood Transfus. 2018, 34, 398–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elenkov, I.J. Glucocorticoids and the Th1/Th2 balance. Ann. N. Y. Acad. Sci. 2004, 1024, 138–146. [Google Scholar] [CrossRef]
- Giles, A.J.; Hutchinson, M.-K.; Sonnemann, H.M.; Jung, J.; Fecci, P.E.; Ratnam, N.M.; Zhang, W.; Song, H.; Bailey, R.; Davis, D.; et al. Dexamethasone-induced immunosuppression: Mechanisms and implications for immunotherapy. J. Immunother. Cancer 2018, 6, 51. [Google Scholar] [CrossRef]
- Gett, A.V.; Hodgkin, P.D. Cell division regulates the T cell cytokine repertoire, revealing a mechanism underlying immune class regulation. Proc. Natl. Acad. Sci. USA 1998, 95, 9488–9493. [Google Scholar] [CrossRef] [Green Version]
- Norbiato, G.; Bevilacqua, M.; Vago, T.; Taddei, A.; Clerici, M. Glucocorticoids and the immune function in the human immunodeficiency virus infection: A study in hypercortisolemic and cortisol-resistant patients. J. Clin. Endocrinol. Metab. 1997, 82, 3260–3263. [Google Scholar] [CrossRef] [Green Version]
- Yamada, H.; Nakashima, Y.; Okazaki, K.; Mawatari, T.; Fukushi, J.I.; Kaibara, N.; Hori, A.; Iwamoto, Y.; Yoshikai, Y. Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 1299–1304. [Google Scholar] [CrossRef]
- Franchimont, D.; Galon, J.; Gadina, M.; Visconti, R.; Zhou, Y.; Aringer, M.; Frucht, D.M.; Chrousos, G.P.; O’Shea, J.J. Inhibition of Th1 immune response by glucocorticoids: Dexamethasone selectively inhibits IL-12-induced Stat4 phosphorylation in T lymphocytes. J. Immunol. 2000, 164, 1768–1774. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.H.; Hassan, A. Dexamethasone for the Treatment of Coronavirus Disease (COVID-19): A Review. SN Compr. Clin. Med. 2020, 2, 2637–2646. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- The RECOVERY Collaborative Group. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Sterne, J.A.; Murthy, S.; Diaz, J.V.; Slutsky, A.S.; Villar, J.; Angus, D.C.; Annane, D.; Azevedo, L.C.; Berwanger, O.; Cavalcanti, A.B.; et al. Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-analysis. JAMA 2020, 324, 1330–1341. [Google Scholar] [CrossRef]
- Villar, J.; Ferrando, C.; Martínez, D.; Ambrós, A.; Muñoz, T.; Soler, J.A.; Aguilar, G.; Alba, F.; González-Higueras, E.; Conesa, L.A.; et al. Dexamethasone treatment for the acute respiratory distress syndrome: A multicentre, randomised controlled trial. Lancet Respir. Med. 2020, 8, 267–276. [Google Scholar] [CrossRef]
- Alhazzani, W.; Møller, M.H.; Arabi, Y.M.; Loeb, M.; Gong, M.N.; Fan, E.; Oczkowski, S.; Levy, M.M.; Derde, L.; Dzierba, A.; et al. Surviving Sepsis Campaign: Guidelines on the Management of Critically Ill Adults with Coronavirus Disease 2019 (COVID-19). Crit. Care Med. 2020, 48, e440–e469. [Google Scholar] [CrossRef]
- Kovacs, W.J. To B or not to B? Glucocorticoid impact on B lymphocyte fate and function. Endocrinology 2014, 155, 339–342. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Tang, J.; Ye, C.; Dong, L. The immunology of COVID-19: Is immune modulation an option for treatment? Lancet Rheumatol. 2020, 2, e428–e436. [Google Scholar] [CrossRef]
- Ai, F.; Zhao, G.; Lv, W.; Liu, B.; Lin, J. Dexamethasone induces aberrant macrophage immune function and apoptosis. Oncol. Rep. 2020, 43, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deborska-Materkowska, D.; Perkowska-Ptasinska, A.; Sadowska-Jakubowicz, A.; Gozdowska, J.; Ciszek, M.; Pazik, J.; Ostaszewska, A.; Kosieradzki, M.; Nowak, J.; Durlik, M. Killer Immunoglobulin-Like Receptor 2DS2 (KIR2DS2), KIR2DL2-HLA-C1, and KIR2DL3 as Genetic Markers for Stratifying the Risk of Cytomegalovirus Infection in Kidney Transplant Recipients. Int. J. Mol. Sci. 2019, 20, 546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bona, D.; Aiello, A.; Colomba, C.; Bilancia, M.; Accardi, G.; Rubino, R.; Giannitrapani, L.; Tuttolomondo, A.; Cascio, A.; Caiaffa, M.F.; et al. KIR2DL3 and the KIR ligand groups HLA-A-Bw4 and HLA-C2 predict the outcome of hepatitis B virus infection. J. Viral Hepat. 2017, 24, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Colomba, C.; Di Bona, D.; Casuccio, A.; Di Raimondo, D.; Clemente, G.; Arnao, V.; Pecoraro, R.; Ragonese, P.; Aiello, A.; et al. HLA and killer cell immunoglobulin-like receptor (KIRs) genotyping in patients with acute viral encephalitis. Oncotarget 2018, 9, 17523–17532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttolomondo, A.; Di Raimondo, D.; Vasto, S.; Casuccio, A.; Colomba, C.; Norrito, R.L.; Di Bona, D.; Arnao, V.; Siciliano, L.; Cascio, A.; et al. Protective and causative killer Ig-like receptor (KIR) and metalloproteinase genetic patterns associated with Herpes simplex virus 1 (HSV-1) encephalitis occurrence. J. Neuroimmunol. 2020, 344, 577241. [Google Scholar] [CrossRef]
- Danson, S.J.; Conner, J.; Edwards, J.G.; Blyth, K.G.; Fisher, P.M.; Muthana, M.; Salawu, A.; Taylor, F.; Hodgkinson, E.; Joyce, P.; et al. Oncolytic herpesvirus therapy for mesothelioma—A phase I/IIa trial of intrapleural administration of HSV1716. Lung Cancer 2020, 150, 145–151. [Google Scholar] [CrossRef]
- Howard, F.; Conner, J.; Danson, S.; Muthana, M. Inconsistencies in Modeling the Efficacy of the Oncolytic Virus HSV1716 Reveal Potential Predictive Biomarkers for Tolerability. Front. Mol. Biosci. 2022, 9, 559. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Feature | Virus | Effect on Host | Refs. |
|---|---|---|---|
| Host genetic susceptibility | |||
| TNF-α (−308) GG genotype IL-10 (-592/-819/-1082) CCA/ATA genotype IL-10 (-592/-819/-1082) ATA/ATG genotype | Dengue | Development of severe dengue in Sri Lankan patients. Risk factor to developing DHF. Protective factor from DHF. | [16] |
| G6PD gene | Dengue, coronavirus, enterovirus | Deficiency enhances viral infection. | [17,18,19] |
| A117V polymorphism in the NS2A | Zika | Increased virulence by reducing host innate immune responses and viral-induced apoptosis in vitro. | [20] |
| HLA DRB1*11 | HCV | Protects from disease progression. | [21] |
| HLA*0405, HLA DRB1*0301, DQB1*0201 | HCV | Viral persistence and chronic infection. | [22] |
| IFIH1 | RSV | Deficient individuals unable to induce IFN-β, rendering them susceptible to infection. | [23] |
| Q421X | IAV | Impaired IFN-α production causes life-threatening condition. | [24] |
| Ageing | |||
| Axl, Mertk | WNV | Age-related upregulation of regulatory receptors facilitates viral uptake by increasing blood–brain barrier permeability. | [25] |
| T-cell defects | WNV | Insufficient number and quality of effector antiviral T-cells underlie age-related susceptibility to WNV. | [26] |
| Histone modifications | IAV | Age-associated altered histone expression decreases IFN production by myeloid DCs. | [27] |
| miR-181a deficiency in T-cells | WNV | Hallmark of ageing. Impairs T-cell expansion, viral clearance, and recall response. | [28] |
| Ethnicity | |||
| IFN | HCV | IFN effectiveness in blocking viral production significantly greater in White versus African-American patients. | [29] |
| ACE2 | COVID-19 | ACE2 (receptor for cellular entry) expression significantly higher among Asians compared to African-Americans and Caucasians. | [30] |
| Nab | Rubella | Individuals of African descent have significantly higher rubella-specific NAb levels than European or Hispanic individuals. | [31] |
| Co-morbidities | |||
| Obesity | Influenza H1N1 | Decreased CD8+ T-cell activation results in inability to mount protective immune response | [32] |
| Asthma | IAV | Increased susceptibility to heterologous secondary influenza due to defective mucosal antibody responses. | [33] |
| Cancer (melanoma/RCC) | Tumour antigen-specific Th2-type polarisation of CD4+ T-cell responses in the peripheral blood of patients with RCC or melanoma. | [34] | |
| Type 2 airway disorders (allergic asthma, allergic rhinitis, CRSwNP) | HRV16 | Type 2 cytokines increase susceptibility to viral infection in airways via changing the epithelial structure and production of interferons. A Th2 bias induces a deficit in defending the mucosa against viral and bacterial infections. | [35] |
| Evidence for Th Polarisation | Ref. | |
|---|---|---|
| Th1 | ||
| Atherosclerosis | CD4+ T-cells dominate atherosclerotic plaques. Increase IFN-γ and IL-2, IL-12, IL-18. | [70] |
| Rheumatoid arthritis | Increase in IFN-γ+CD4+ T-cells in peripheral blood and IFN-γ and TNF-α expression. Reduction in IL-6 and IL10 expression. | [105] |
| Type I diabetes | High IFN-γ expression drives persistent signal in pancreatic beta cells. | [106] |
| Multiple sclerosis | IFN-γ-producing Th1 cells most frequent Th cell subset in the CNS. | [107] |
| Parkinson’s | PD patients more Th1 cells and fewer Treg cells. CD4+ T-cells mediate brain inflammation. | [108] [109] |
| Th2 | ||
| Asthma | Production of Th2 cytokines IL-4, IL-13, IL-5, increased production of IgE by B-cells. Genes that enhanced Th2 polarisation (IL17RB) and Th2 cytokine (IL-25) production were upregulated in asthma. | [110] [111] |
| Ulcerative colitis | Overexpression of Bcl2L12 by CD4+ T-cells upregulates Th2 responses and downregulates Th2 ell apoptosis. | [112] |
| Chronic fatigue syndrome | Shift from Th1 to Th2 profile correlated with illness parameters including increase in IL-4 and reduced natural killer cell cytotoxicity. | [113] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Howard, F.H.N.; Kwan, A.; Winder, N.; Mughal, A.; Collado-Rojas, C.; Muthana, M. Understanding Immune Responses to Viruses—Do Underlying Th1/Th2 Cell Biases Predict Outcome? Viruses 2022, 14, 1493. https://doi.org/10.3390/v14071493
Howard FHN, Kwan A, Winder N, Mughal A, Collado-Rojas C, Muthana M. Understanding Immune Responses to Viruses—Do Underlying Th1/Th2 Cell Biases Predict Outcome? Viruses. 2022; 14(7):1493. https://doi.org/10.3390/v14071493
Chicago/Turabian StyleHoward, Faith H. N., Amy Kwan, Natalie Winder, Amina Mughal, Cristal Collado-Rojas, and Munitta Muthana. 2022. "Understanding Immune Responses to Viruses—Do Underlying Th1/Th2 Cell Biases Predict Outcome?" Viruses 14, no. 7: 1493. https://doi.org/10.3390/v14071493