
Inducible miR-150 Inhibits Porcine Reproductive and Respiratory Syndrome Virus Replication by Targeting Viral Genome and Suppressor of Cytokine Signaling 1

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Small RNA Deep Sequencing
2.3. Indirect Immunofluorescence Antibody (IFA) Assay
2.4. Transfection of miRNA Mimic and Viral Infections
2.5. Inhibition of Signaling Transduction Pathways
2.6. RNA Extraction and Real-Time PCR
2.7. Western Blot
2.8. RNA Immunoprecipitation Assay
2.9. Plasmid Construction and Dual Luciferase Reporter Assays
2.10. Chromatin Immunoprecipitation (ChIP) Assay
2.11. Statistical Analysis
3. Results
3.1. miR-150 Is Induced by PRRSV Infection
3.2. PRRSV Induces miR-150 Expression by Activating the PKC/JNK/c-Jun Pathway
3.3. c-Jun Is Crucial for PRRSV to Activate the miR-150 Promoter
3.4. miR-150 Inhibits PRRSV Replication
3.5. miR-150 Directly Targets the PRRSV Genome
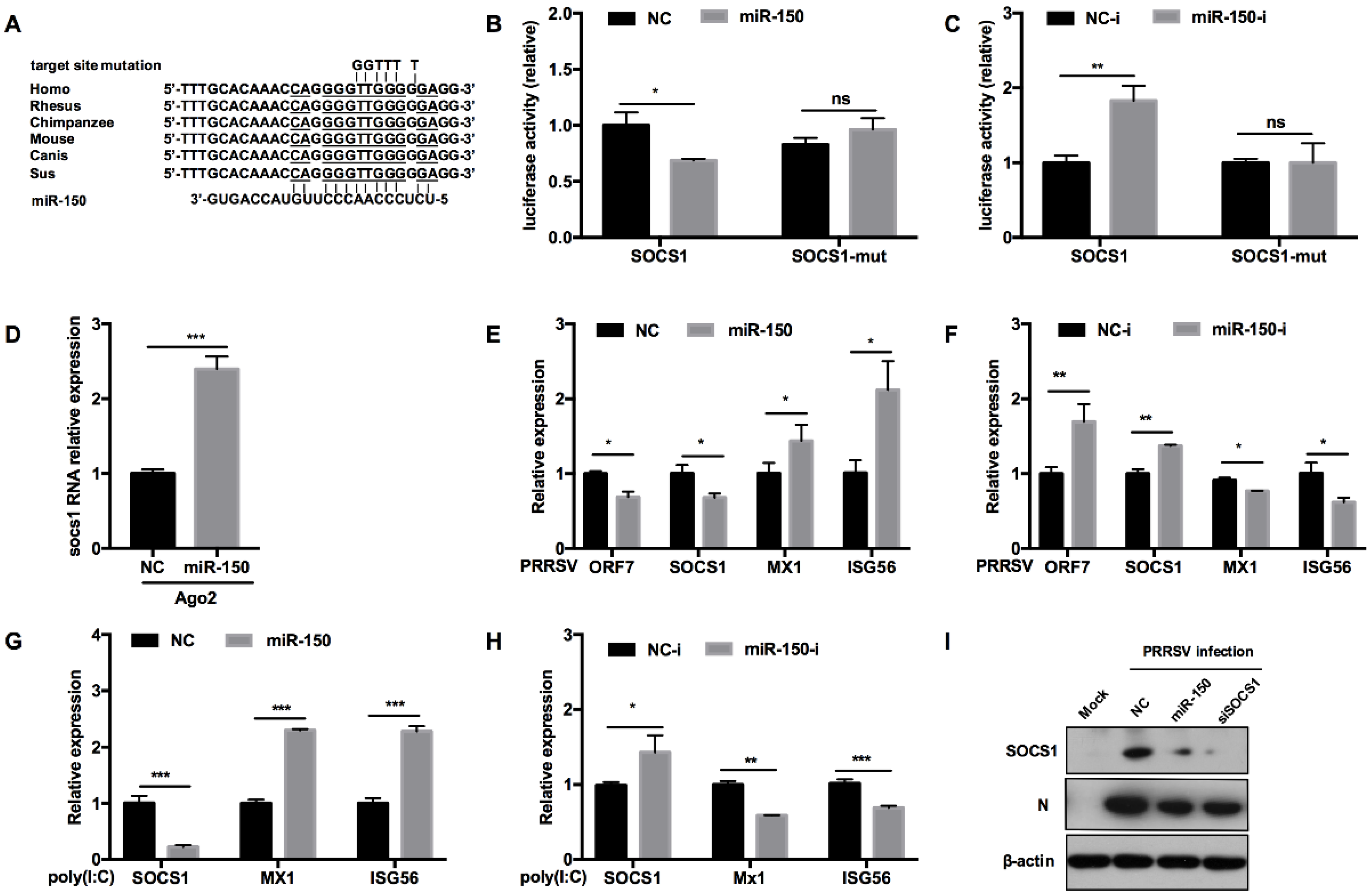
3.6. miR-150 Facilitates Type I IFN Responses by Targeting SOCS1
3.7. miR-150 Has Inhibition of Porcine RNA Virus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lunney, J.K.; Fang, Y.; Ladinig, A.; Chen, N.; Li, Y.; Rowland, B.; Renukaradhya, G.J. Porcine Reproductive and Respiratory Syndrome Virus (PRRSV): Pathogenesis and Interaction with the Immune System. Annu. Rev. Anim. Biosci. 2016, 4, 129–154. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Chen, X.X.; Li, R.; Qiao, S.; Zhang, G. The prevalent status and genetic diversity of porcine reproductive and respiratory syndrome virus in China: A molecular epidemiological perspective. Virol. J. 2018, 15, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappes, M.A.; Faaberg, K.S. PRRSV structure, replication and recombination: Origin of phenotype and genotype diversity. Virology 2015, 479-480, 475–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaner-Tarbes, S.; del Portillo, H.A.; Montoya, M.; Fraile, L. Key Gaps in the Knowledge of the Porcine Respiratory Reproductive Syndrome Virus (PRRSV). Front. Vet. Sci. 2019, 6, 38. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, Y.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; Chen, S.; et al. Structures and Functions of the 3′ Untranslated Regions of Positive-Sense Single-Stranded RNA Viruses Infecting Humans and Animals. Front. Cell Infect. Microbiol. 2020, 10, 453. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, C.; Tan, F.; Gao, F.; Liu, P.; Qin, A.; Yuan, S. Identification of dispensable nucleotide sequence in 3′ untranslated region of porcine reproductive and respiratory syndrome virus. Virus Res. 2010, 154, 38–47. [Google Scholar] [CrossRef]
- Xie, S.; Chen, X.X.; Qiao, S.; Li, R.; Sun, Y.; Xia, S.; Wang, L.J.; Luo, X.; Deng, R.; Zhou, E.M.; et al. Identification of the RNA Pseudoknot within the 3′ End of the Porcine Reproductive and Respiratory Syndrome Virus Genome as a Pathogen-Associated Molecular Pattern To Activate Antiviral Signaling via RIG-I and Toll-Like Receptor 3. J. Virol. 2018, 92, e00097-18. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Liu, C.; Liu, P.; Yao, H.; Wei, Z.; Lu, J.; Tong, G.; Gao, F.; Yuan, S. Conserved nucleotides in the terminus of the 3′ UTR region are important for the replication and infectivity of porcine reproductive and respiratory syndrome virus. Arch. Virol. 2013, 158, 1719–1732. [Google Scholar] [CrossRef]
- Han, J.; Zhou, L.; Ge, X.; Guo, X.; Yang, H. Pathogenesis and control of the Chinese highly pathogenic porcine reproductive and respiratory syndrome virus. Vet. Microbiol. 2017, 209, 30–47. [Google Scholar] [CrossRef]
- Zhou, Z.; Ni, J.; Cao, Z.; Han, X.; Xia, Y.; Zi, Z.; Ning, K.; Liu, Q.; Cai, L.; Qiu, P.; et al. The epidemic status and genetic diversity of 14 highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV) isolates from China in 2009. Vet. Microbiol. 2011, 150, 257–269. [Google Scholar] [CrossRef]
- Li, B.; Fang, L.; Guo, X.; Gao, J.; Song, T.; Bi, J.; He, K.; Chen, H.; Xiao, S. Epidemiology and evolutionary characteristics of the porcine reproductive and respiratory syndrome virus in China between 2006 and 2010. J. Clin. Microbiol. 2011, 49, 3175–3183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corzo, C.A.; Mondaca, E.; Wayne, S.; Torremorell, M.; Dee, S.; Davies, P.; Morrison, R.B. Control and elimination of porcine reproductive and respiratory syndrome virus. Virus Res. 2010, 154, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Fay, E.J.; Langlois, R.A. MicroRNA-Attenuated Virus Vaccines. Noncoding RNA 2018, 4, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameres, S.L.; Zamore, P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, D. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef]
- Du, T.; Nan, Y.; Xiao, S.; Zhao, Q.; Zhou, E.M. Antiviral Strategies against PRRSV Infection. Trends Microbiol. 2017, 25, 968–979. [Google Scholar] [CrossRef]
- Liu, F.; Du, Y.; Feng, W.H. New perspective of host microRNAs in the control of PRRSV infection. Vet. Microbiol. 2017, 209, 48–56. [Google Scholar] [CrossRef]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, C.S.; Ganem, D. MicroRNAs and viral infection. Mol. Cell 2005, 20, 3–7. [Google Scholar] [CrossRef]
- Trobaugh, D.W.; Klimstra, W.B. MicroRNA Regulation of RNA Virus Replication and Pathogenesis. Trends Mol. Med. 2017, 23, 80–93. [Google Scholar] [CrossRef]
- Chow, K.T.; Gale, M.; Loo, Y. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 2005, 23, 307–336. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Huang, C.; Yang, Q.; Gao, L.; Liu, H.C.; Tang, J.; Feng, W.H. MicroRNA-30c Modulates Type I IFN Responses To Facilitate Porcine Reproductive and Respiratory Syndrome Virus Infection by Targeting JAK1. J. Immunol. 2016, 196, 2272–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, X.; Shi, X.; Zhang, X.; Chen, J.; Fan, X.; Yang, Y.; Wang, L.; Wang, A.; Deng, R.; Zhou, E.M.; et al. miR-382-5p promotes porcine reproductive and respiratory syndrome virus (PRRSV) replication by negatively regulating the induction of type I interferon. FASEB J. 2020, 34, 4497–4511. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Shi, X.; Zhang, X.; Wang, A.; Wang, L.; Yang, Y.; Deng, R.; Zhang, G.P. MicroRNA 373 Facilitates the Replication of Porcine Reproductive and Respiratory Syndrome Virus by Its Negative Regulation of Type I Interferon Induction. J. Virol. 2017, 91, e01311-16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, L.; Pan, Y.; Gao, J.; Xu, Y.; Li, X.; Tian, Z.; Chen, H.; Wang, Y. Downregulation of miR-218 by porcine reproductive and respiratory syndrome virus facilitates viral replication via inhibition of type I interferon responses. J. Biol. Chem. 2021, 296, 100683. [Google Scholar] [CrossRef]
- Zhang, Q.; Guo, X.K.; Gao, L.; Huang, C.; Li, N.; Jia, X.; Liu, W.; Feng, W.H. MicroRNA-23 inhibits PRRSV replication by directly targeting PRRSV RNA and possibly by upregulating type I interferons. Virology 2014, 450–451, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wei, Z.; Zhou, Y.; Gao, Y.; Jiang, Y.; Yu, L.; Zheng, H.; Tong, W.; Yang, S.; Zheng, H.; et al. Host miR-26a suppresses replication of porcine reproductive and respiratory syndrome virus by upregulating type I interferons. Virus Res. 2015, 195, 86–94. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, H.; Suo, X.; Zheng, S.; Feng, W.H. Increase of CD163 but not sialoadhesin on cultured peripheral blood monocytes is coordinated with enhanced susceptibility to porcine reproductive and respiratory syndrome virus infection. Vet. Immunol. Immunopathol. 2011, 141, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, M.A. Determination of 50% endpoint titer using a simple formula. World J. Virol. 2016, 5, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Ramani, R.; Suraju, M.O. Polyphenol compounds and PKC signaling. Biochim Biophys Acta 2016, 1860, 2107–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakov, N. Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin. Cancer Biol. 2018, 48, 36–52. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q.; Liu, Y.; Luo, Z.; Kang, L.; Qu, J.; Liu, W.; Xia, X.; Liu, Y.; Wu, K.; et al. Hepatitis C virus activates Bcl-2 and MMP-2 expression through multiple cellular signaling pathways. J. Virol. 2012, 86, 12531–12543. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Luo, A.; Zhou, C.; Ding, F.; Wu, M.; Zhan, Q.; Liu, Z. Differentiation-associated genes regulated by TPA-induced c-Jun expression via a PKC/JNK pathway in KYSE450 cells. Biochem. Biophys. Res. Commun. 2006, 342, 286–292. [Google Scholar] [CrossRef]
- Meng, Q.; Xia, Y. c-Jun, at the crossroad of the signaling network. Protein Cell 2011, 2, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Chen, X.X.; Qiao, S.; Li, R.; Xie, S.; Zhou, X.; Deng, R.; Zhou, E.M.; Zhang, G. Porcine Reproductive and Respiratory Syndrome Virus Enhances Self-Replication via AP-1-Dependent Induction of SOCS1. J. Immunol. 2020, 204, 394–407. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Nauwynck, H.J.; Pensaert, M.B. Effects of origin and state of differentiation and activation of monocytes:macrophages on their susceptibility to porcine reproductive and respiratory syndrome virus (PRRSV). Achives Virol. 1997, 142, 2483–2497. [Google Scholar]
- Fan, Y.; Zhu, L.; Sun, X.; Lyu, W.; Xu, L.; Yin, Y.; Zhao, J.; Huang, J.; Den, Y.; Jiang, Z.; et al. Exploring the tissue tropism of pseudorabies virus based on miRNA level analysis. BMC Microbiol. 2019, 19, 125. [Google Scholar] [CrossRef]
- Guo, X.K.; Zhang, Q.; Gao, L.; Li, N.; Chen, X.X.; Feng, W.H. Increasing expression of microRNA 181 inhibits porcine reproductive and respiratory syndrome virus replication and has implications for controlling virus infection. J. Virol 2013, 87, 1159–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunden, R.D.; Khan, J.Q.; Ghezelbash, S.; Wilson, J.A. The Role of the Liver-Specific microRNA, miRNA-122 in the HCV Replication Cycle. Int. J. Mol. Sci. 2020, 21, 5677. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [PubMed]
- Creugny, A.; Fender, A.; Pfeffer, S. Regulation of primary microRNA processing. FEBS Lett. 2018, 592, 1980–1996. [Google Scholar] [CrossRef] [PubMed]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef]
- Liu, X.; Gao, L.; Zhao, Q.; Wang, X.; Yang, C.; Bi, J.; Yang, R.; Jin, X.; Lan, R.; Cui, R.; et al. Inhibition of porcine reproductive and respiratory syndrome virus by PKC inhibitor dequalinium chloride in vitro. Vet. Microbiol. 2020, 251, 108913. [Google Scholar] [CrossRef]
- Meineke, R.; Rimmelzwaan, G.F.; Elbahesh, H. Influenza Virus Infections and Cellular Kinases. Viruses 2019, 11, 171. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Guo, X.K.; Bi, Y.; Zhu, Y.; Feng, W.H. PKCdelta is required for porcine reproductive and respiratory syndrome virus replication. Virology 2014, 468-470, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Rosse, C.; Linch, M.; Kermorgant, S.; Cameron, A.J.; Boeckeler, K.; Parker, P.J. PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 2010, 11, 103–112. [Google Scholar] [CrossRef]
- Gao, L.; Guo, X.K.; Wang, L.; Zhang, Q.; Li, N.; Chen, X.X.; Wang, Y.; Feng, W.H. MicroRNA 181 suppresses porcine reproductive and respiratory syndrome virus (PRRSV) infection by targeting PRRSV receptor CD163. J. Virol. 2013, 87, 8808–8812. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Huang, K.; Chen, Y.; Huang, Z.; Zhang, Y.; Leng, C.; Liu, Y.; Shi, J.; Xiao, S.; Yao, L. MicroRNA ssc-miR-124a exhibits antiviral activity against porcine reproductive and respiratory syndrome virus via suppression of host genes CD163. Vet. Microbiol. 2021, 261, 109216. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Peng, X.; Zhou, A.; Qiao, M.; Wu, H.; Xiao, H.; Liu, G.; Zheng, X.; Zhang, S.; Mei, S. MiR-506 inhibits PRRSV replication in MARC-145 cells via CD151. Mol. Cell Biochem. 2014, 394, 275–281. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Qu, Y.; Zhang, Y.; Huang, J.; Gao, X.; Huang, C.; Luo, G.; Liu, Q.; Liu, M.; Xu, D. Mir-331-3p Inhibits PRRSV-2 Replication and Lung Injury by Targeting PRRSV-2 ORF1b and Porcine TNF-alpha. Front. Immunol. 2020, 11, 547144. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Du, T.; Wang, X.; Ni, H.; Yan, Y.; Li, N.; Zhang, C.; Zhang, A.; Gao, J.; Liu, H.; et al. MiR-22 promotes porcine reproductive and respiratory syndrome virus replication by targeting the host factor HO-1. Vet. Microbiol 2016, 192, 226–230. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, S.; Cui, Z.; Li, W.; Xu, P.; Liu, H.; Miao, X.; Chen, Y.; Han, F.; Zhang, H.; et al. MicroRNA-376b-3p Promotes Porcine Reproductive and Respiratory Syndrome Virus Replication by Targeting Viral Restriction Factor TRIM22. J. Virol. 2022, 96, e0159721. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef]
- Krebs, D.L.; Hilton, D.J. SOCS Proteins: Negative Regulators of Cytokine Signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef]
- Huang, S.; Liu, K.; Cheng, A.; Wang, M.; Cui, M.; Huang, J.; Zhu, D.; Chen, S.; Liu, M.; Zhao, X.; et al. SOCS Proteins Participate in the Regulation of Innate Immune Response Caused by Viruses. Front. Immunol. 2020, 11, 558341. [Google Scholar] [CrossRef]
- Chen, R.F.; Yang, K.D.; Lee, I.K.; Liu, J.W.; Huang, C.H.; Lin, C.Y.; Chen, Y.H.; Chen, C.L.; Wang, L. Augmented miR-150 expression associated with depressed SOCS1 expression involved in dengue haemorrhagic fever. J. Infect. 2014, 69, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Hasni, S.A.; Perez, P.; Tandon, M.; Jang, S.-I.; Zheng, C.; Kopp, J.B.; Austin, H.; Balow, J.E.; Alevizos, I.; et al. miR-150 Promotes Renal Fibrosis in Lupus Nephritis by Downregulating SOCS1. J. Am. Soc. Nephrol. 2013, 24, 1073–1087. [Google Scholar] [CrossRef] [Green Version]
- Luan, J.; Fu, J.; Wang, D.; Jiao, C.; Cui, X.; Chen, C.; Liu, D.; Zhang, Y.; Wang, Y.; Yuen, P.S.T.; et al. miR-150-Based RNA Interference Attenuates Tubulointerstitial Fibrosis through the SOCS1/JAK/STAT Pathway In Vivo and In Vitro. Mol. Ther. Nucleic Acids 2020, 22, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ye, L.; Zhou, Y.; Liu, M.Q.; Zhou, D.J.; Ho, W.Z. Inhibition of anti-HIV microRNA expression: A mechanism for opioid-mediated enhancement of HIV infection of monocytes. Am. J. Pathol. 2011, 178, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, G.; Yao, Z.Q.; Moorman, J.P.; Ning, S. MicroRNA regulation of viral immunity, latency, and carcinogenesis of selected tumor viruses and HIV. Rev. Med. Virol. 2015, 25, 320–341. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef]
- Dubey, R.C.; Alam, N.B.; Gaur, R. miR-150-mediated increase in glucose uptake in HIV-infected cells. J. Med. Virol. 2021, 93, 6377–6382. [Google Scholar] [CrossRef]
- He, T.; Feng, G.; Chen, H.; Wang, L.; Wang, Y. Identification of host encoded microRNAs interacting with novel swine-origin influenza A (H1N1) virus and swine influenza virus. Bioinformation 2009, 4, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Moran, J.; Ramirez-Martinez, G.; Jimenez-Alvarez, L.; Cruz, A.; Perez-Patrigeon, S.; Hidalgo, A.; Orozco, L.; Martinez, A.; Padilla-Noriega, L.; Avila-Moreno, F.; et al. Circulating levels of miR-150 are associated with poorer outcomes of A/H1N1 infection. Exp. Mol. Pathol. 2015, 99, 253–261. [Google Scholar] [CrossRef]
- Akula, S.M.; Bolin, P.; Cook, P.P. Cellular miR-150-5p may have a crucial role to play in the biology of SARS-CoV-2 infection by regulating nsp10 gene. RNA Biol. 2022, 19, 1–11. [Google Scholar] [CrossRef]
- Huang, X.L.; Zhang, L.; Li, J.P.; Wang, Y.J.; Duan, Y.; Wang, J. MicroRNA-150: A potential regulator in pathogens infection and autoimmune diseases. Autoimmunity 2015, 48, 503–510. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Zhang, X.; Yao, Y.; Zhu, Y.; Zheng, X.; Liu, F.; Feng, W. Inducible miR-150 Inhibits Porcine Reproductive and Respiratory Syndrome Virus Replication by Targeting Viral Genome and Suppressor of Cytokine Signaling 1. Viruses 2022, 14, 1485. https://doi.org/10.3390/v14071485
Li S, Zhang X, Yao Y, Zhu Y, Zheng X, Liu F, Feng W. Inducible miR-150 Inhibits Porcine Reproductive and Respiratory Syndrome Virus Replication by Targeting Viral Genome and Suppressor of Cytokine Signaling 1. Viruses. 2022; 14(7):1485. https://doi.org/10.3390/v14071485
Chicago/Turabian StyleLi, Sihan, Xuan Zhang, Yao Yao, Yingqi Zhu, Xiaojie Zheng, Fang Liu, and Wenhai Feng. 2022. "Inducible miR-150 Inhibits Porcine Reproductive and Respiratory Syndrome Virus Replication by Targeting Viral Genome and Suppressor of Cytokine Signaling 1" Viruses 14, no. 7: 1485. https://doi.org/10.3390/v14071485