
A Comparison of Pseudorabies Virus Latency to Other α-Herpesvirinae Subfamily Members

Abstract
:1. Introduction
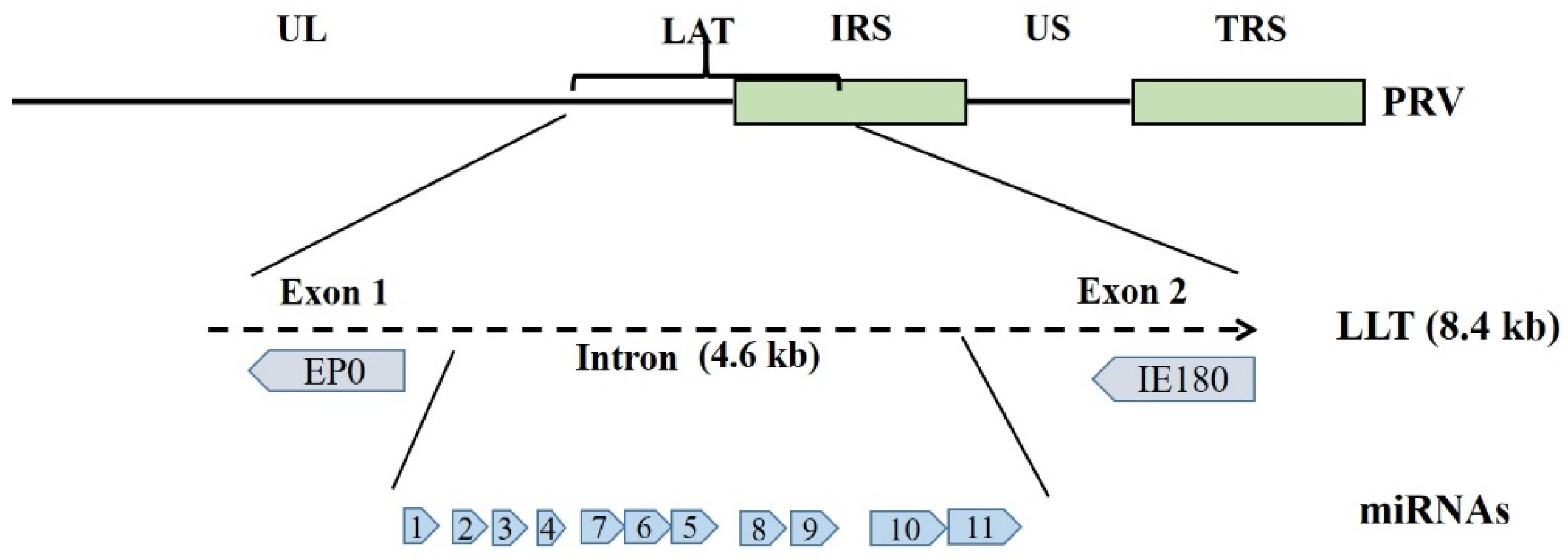
2. The Viral Latency-Associated Transcripts in PRV and Other Alphaherpesviruses Latency
3. Viral Non-Coding RNAs in PRV and Other Alphaherpesviruses Latency
4. The Viral Proteins Directly and Indirectly Participated in Alphaherpesviruses Latency
5. Immune Regulation of PRV and Other Alphaherpesviruses Latency
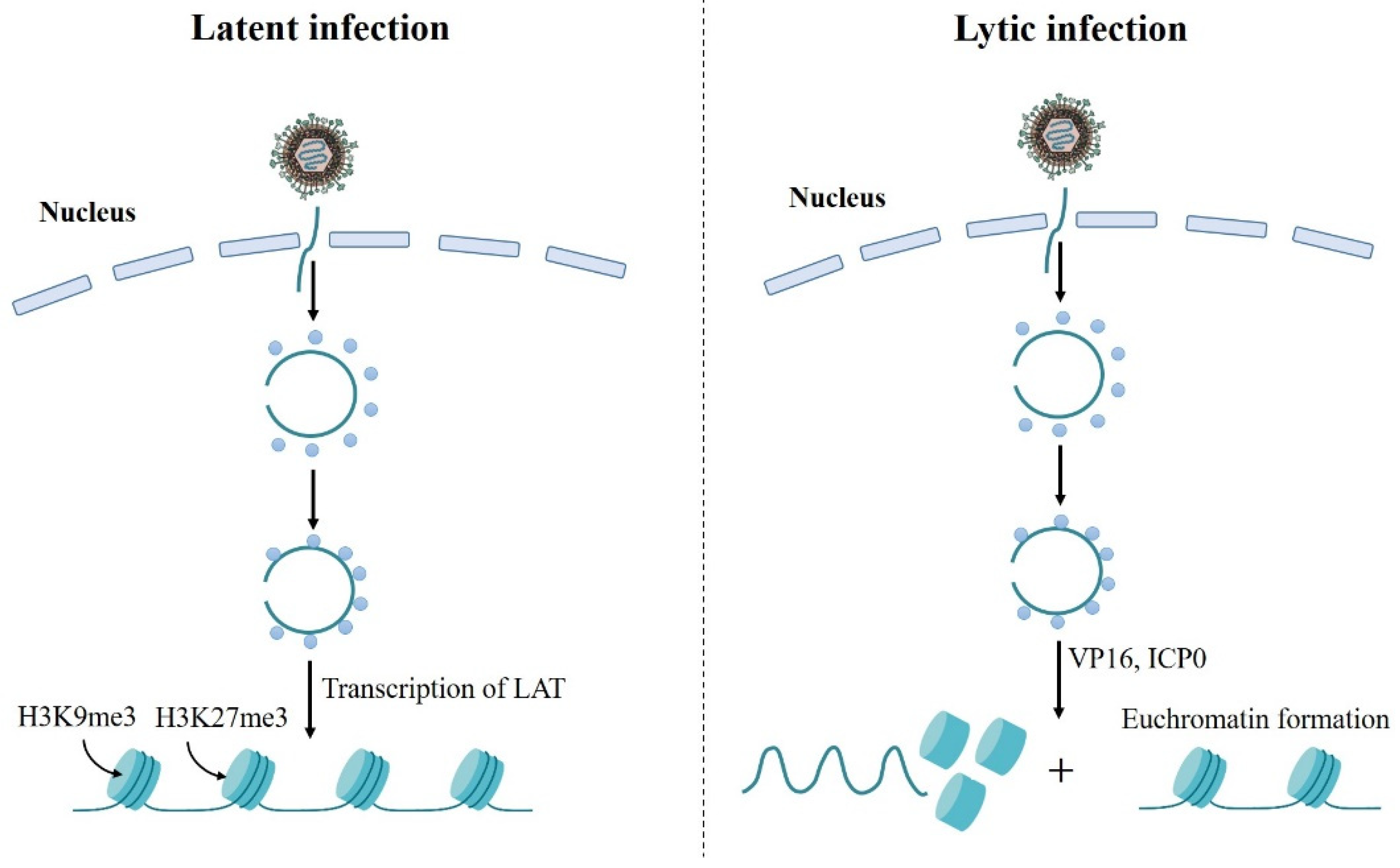
6. Chromatin Regulation of Alphaherpesviruses Latency
7. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.Y.; Wang, X.J.; Xie, C.H.; Ding, S.F.; Yang, H.N.; Guo, S.B.; Li, J.X.; Qin, L.Z.; Ban, F.G.; Wang, D.F.; et al. A novel human acute encephalitis caused by pseudorabies virus variant strain. Clin. Infect. Dis. 2021, 73, e3690–e3700. [Google Scholar] [CrossRef] [PubMed]
- An, T.Q.; Peng, J.M.; Tian, Z.J.; Zhao, H.Y.; Li, N.; Liu, Y.M.; Chen, J.Z.; Leng, C.L.; Sun, Y.; Chang, D.; et al. Pseudorabies virus variant in Bartha-K61–vaccinated pigs, China, 2012. Emerg. Infect. Dis. 2013, 19, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhou, Z.; Hu, D.; Zhang, Q.; Han, T.; Li, X.; Gu, X.; Yuan, L.; Zhang, S.; Wang, B.; et al. Pathogenic pseudorabies virus, China, 2012. Emerg. Infect. Dis. 2014, 20, 102–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Luo, Y.Z.; Wang, C.H.; Yuan, J.; Li, N.; Song, K.; Qiu, H.J. Control of swine pseudorabies in China: Opportunities and limitations. Vet. Microbiol. 2016, 183, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K. Detection of pseudorabies virus transcripts in trigeminal ganglia of latently infected swine. J. Virol. 1989, 63, 2908–2913. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Schnitzlein, W.M.; Scherba, G. Identification of the pseudorabies virus promoter required for latency-associated transcript gene expression in the natural host. J. Virol. 2000, 74, 6333–6338. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.K. DNA nucleotide sequence analysis of the immediate-early gene of pseudorabies virus. Nucleic. Acids Res. 1989, 17, 4637–4646. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.K. Cloning of the latency gene and the early protein 0 gene of pseudorabies virus. J. Virol. 1991, 65, 5260–5271. [Google Scholar] [CrossRef] [Green Version]
- Maes, R.K.; Sussman, M.D.; Vilnis, A.; Thacker, B.J. Recent developments in latency and recombination of Aujeszky’s disease (pseudorabies) virus. Vet. Microbiol. 1997, 55, 13–27. [Google Scholar] [CrossRef]
- Priola, S.A.; Stevens, J.G. The 5′ and 3′ limits of transcription in the pseudorabies virus latency associated transcription unit. Virology 1991, 182, 852–856. [Google Scholar] [CrossRef]
- Mahjoub, N.; Dhorne-Pollet, S.; Fuchs, W.; Endale Ahanda, M.L.; Lange, E.; Klupp, B.; Arya, A.; Loveland, J.E.; Lefevre, F.; Mettenleiter, T.C.; et al. A 2.5-kilobase deletion containing a cluster of nine microRNAs in the latency-associated-transcript locus of the pseudorabies virus affects the host response of porcine trigeminal ganglia during established latency. J. Virol. 2015, 89, 428–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anselmo, A.; Flori, L.; Jaffrezic, F.; Rutigliano, T.; Cecere, M.; Cortes-Perez, N.; Lefèvre, F.; Rogel-Gaillard, C.; Giuffra, E. Co-expression of host and viral microRNAs in porcine dendritic cells infected by the pseudorabies virus. PLoS ONE 2011, 6, e17374. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.Q.; Chen, D.J.; He, H.B.; Chen, D.S.; Chen, L.L.; Chen, H.C.; Liu, Z.F. Pseudorabies virus infected porcine epithelial cell line generates a diverse set of host microRNAs and a special cluster of viral microRNAs. PLoS ONE 2012, 7, e30988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Scherba, G. Expression of the pseudorabies virus latency associated transcript gene during productive infection of cultured cells. J. Virol. 1999, 73, 9781–9788. [Google Scholar] [CrossRef] [Green Version]
- Perng, G.C.; Dunkel, E.C.; Geary, P.A.; Slanina, S.M.; Ghiasi, H.; Kaiwar, R.; Nesburn, A.B.; Wechsler, S.L. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J. Virol. 1994, 68, 8045–8055. [Google Scholar] [CrossRef] [Green Version]
- Perng, G.C.; Chokephaibulkit, K.; Thompson, R.L.; Sawtell, N.M.; Slanina, S.M.; Ghiasi, H.; Nesburn, A.B.; Wechsler, S.L. The region of the herpes simplex virus type 1 LAT gene that is colinear with the ICP34.5 gene is not involved in spontaneous reactivation. J. Virol. 1996, 70, 282–291. [Google Scholar] [CrossRef] [Green Version]
- Leib, D.A.; Bogard, C.L.; Kosz-Vnenchak, M.; Hicks, K.A.; Coen, D.M.; Knipe, D.M.; Schaffer, P.A. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J. Virol. 1989, 63, 2893–2900. [Google Scholar] [CrossRef] [Green Version]
- Depledge, D.P.; Ouwendijk, W.; Sadaoka, T.; Braspenning, S.E.; Mori, Y.; Cohrs, R.J.; Verjans, G.; Breuer, J. A spliced latency-associated VZV transcript maps antisense to the viral transactivator gene 61. Nat. Commun. 2018, 9, 1167. [Google Scholar] [CrossRef]
- Kennedy, P.G.; Rovnak, J.; Badani, H.; Cohrs, R.J. A comparison of herpes simplex virus type 1 and varicella-zoster virus latency and reactivation. J. Gen. Virol. 2015, 96, 1581–1602. [Google Scholar] [CrossRef]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, M.F.; Jurak, I.; Pesola, J.M.; Boissel, S.; Knipe, D.M.; Coen, D.M. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology 2011, 417, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Brown, D.; Osorio, N.; Hsiang, C.; Li, L.; Chan, L.; BenMohamed, L.; Wechsler, S.L. A herpes simplex virus type 1 mutant disrupted for microRNA H2 with increased neurovirulence and rate of reactivation. J. Neurovirol. 2015, 21, 199–209. [Google Scholar] [CrossRef]
- Feldman, E.R.; Tibbetts, S.A. Emerging roles of herpesvirus microRNAs during in vivo infection and pathogenesis. Curr. Pathobiol. Rep. 2015, 3, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Vitvitskaia, O.; Carpenter, D.; Wechsler, S.L.; Jones, C. Identification of two small RNAs within the first 1.5-kb of the herpes simplex virus type 1-encoded latency-associated transcript. J. Neurovirol. 2008, 14, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Tormanen, K.; Wang, S.; Matundan, H.H.; Yu, J.; Jaggi, U.; Ghiasi, H. Herpes simplex virus 1 small noncoding RNAs 1 and 2 activate the herpesvirus entry mediator promoter. J. Virol. 2022, 96, e0198521. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Silva, M.; Jaber, T.; Vitvitskaia, O.; Li, S.; Henderson, G.; Jones, C. Two small RNAs encoded within the first 1.5 kilobases of the herpes simplex virus type 1 latency-associated transcript can inhibit productive infection and cooperate to inhibit apoptosis. J. Virol. 2009, 83, 9131–9139. [Google Scholar] [CrossRef] [Green Version]
- da Silva, L.F.; Jones, C. Small non-coding RNAs encoded within the herpes simplex virus type 1 latency associated transcript (LAT) cooperate with the retinoic acid inducible gene I (RIG-I) to induce beta-interferon promoter activity and promote cell survival. Virus Res. 2013, 175, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Tormanen, K.; Matundan, H.H.; Wang, S.; Jaggi, U.; Mott, K.R.; Ghiasi, H. Small noncoding RNA (sncRNA1) within the latency-associated transcript modulates herpes simplex virus 1 virulence and the host immune response during acute but not latent infection. J. Virol. 2022, 96, e0005422. [Google Scholar] [CrossRef]
- Gordon, Y.J.; Gilden, D.M.; Becker, Y. HSV-1 thymidine kinase promotes virulence and latency in the mouse. Investig. Ophthalmol. Vis. Sci. 1983, 24, 599–602. [Google Scholar]
- Meignier, B.; Longnecker, R.; Mavromara-Nazos, P.; Sears, A.E.; Roizman, B. Virulence of and establishment of latency by genetically engineered deletion mutants of herpes simplex virus 1. Virology 1988, 162, 251–254. [Google Scholar] [CrossRef]
- Coen, D.M.; Kosz-Vnenchak, M.; Jacobson, J.G.; Leib, D.A.; Bogard, C.L.; Schaffer, P.A.; Tyler, K.L.; Knipe, D.M. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc. Natl. Acad. Sci. USA 1989, 86, 4736–4740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroop, W.G.; Banks, M.C.; Qavi, H.; Chodosh, J.; Brown, S.M. A thymidine kinase deficient HSV-2 strain causes acute keratitis and establishes trigeminal ganglionic latency, but poorly reactivates in vivo. J. Med. Virol. 1994, 43, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.A.; Field, H.J. Experimental reactivation of bovine herpesvirus 1 (BHV-1) by means of corticosteroids in an intranasal rabbit model. Arch. Virol. 1990, 112, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Whetstone, C.A.; Miller, J.M.; Seal, B.S.; Bello, L.J.; Lawrence, W.C. Latency and reactivation of a thymidine kinase-negative bovine herpesvirus 1 deletion mutant. Arch. Virol. 1992, 122, 207–214. [Google Scholar] [CrossRef]
- Ferrari, M.; Mettenleiter, T.C.; Romanelli, M.G.; Cabassi, E.; Corradi, A.; Dal Mas, N.; Silini, R. A comparative study of pseudorabies virus (PRV) strains with defects in thymidine kinase and glycoprotein genes. J. Comp. Pathol. 2000, 123, 152–163. [Google Scholar] [CrossRef]
- Volz, D.M.; Lager, K.M.; Mengeling, W.L. Latency of a thymidine kinase-negative pseudorabies vaccine-virus detected by the polymerase chain reaction. Arch. Virol. 1992, 122, 341–348. [Google Scholar] [CrossRef]
- Jacobs, L. Glycoprotein E of pseudorabies virus and homologous proteins in other alphaherpesvirinae. Arch. Virol. 1994, 137, 209–228. [Google Scholar] [CrossRef]
- Yang, M.; Card, J.P.; Tirabassi, R.S.; Miselis, R.R.; Enquist, L.W. Retrograde, transneuronal spread of pseudorabies virus in defined neuronal circuitry of the rat brain is facilitated by gE mutations that reduce virulence. J. Virol. 1999, 73, 4350–4359. [Google Scholar] [CrossRef] [Green Version]
- DuRaine, G.; Johnson, D.C. Anterograde transport of α-herpesviruses in neuronal axons. Virology 2021, 559, 65–73. [Google Scholar] [CrossRef]
- Cheung, A.K. Latency characteristics of an EPO and LLT mutant of pseudorabies virus. J. Vet. Diagn. Investig. 1996, 8, 112–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldogköi, Z.; Braun, A.; Fodor, I. Replication and virulence of early protein 0 and long latency transcript deficient mutants of the Aujeszky’s disease (pseudorabies) virus. Microbes Infect. 2000, 2, 1321–1328. [Google Scholar] [CrossRef]
- Wang, H.H.; Liu, J.; Li, L.T.; Chen, H.C.; Zhang, W.P.; Liu, Z.F. Typical gene expression profile of pseudorabies virus reactivation from latency in swine trigeminal ganglion. J. Neurovirol. 2020, 26, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Ou, C.J.; Wong, M.L.; Huang, C.; Chang, T.J. Suppression of promoter activity of the LAT gene by IE180 of pseudorabies virus. Virus Genes 2002, 25, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Sinani, D.; Liu, Y.; Jones, C. Analysis of a bovine herpesvirus 1 protein encoded by an alternatively spliced latency related (LR) RNA that is abundantly expressed in latently infected neurons. Virology 2014, 464, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Kutish, G.; Mainprize, T.; Rock, D. Characterization of the latency-related transcriptionally active region of the bovine herpesvirus 1 genome. J. Virol. 1990, 64, 5730–5737. [Google Scholar] [CrossRef] [Green Version]
- Meyer, F.; Perez, S.; Jiang, Y.; Zhou, Y.; Henderson, G.; Jones, C. Identification of a novel protein encoded by the latency-related gene of bovine herpesvirus 1. J. Neurovirol. 2007, 13, 569–578. [Google Scholar] [CrossRef]
- Jiang, Y.; Inman, M.; Zhang, Y.; Posadas, N.A.; Jones, C. A mutation in the latency-related gene of bovine herpesvirus 1 inhibits protein expression of a protein from open reading frame 2 and an adjacent reading frame during productive infection. J. Virol. 2004, 78, 3184–3189. [Google Scholar] [CrossRef] [Green Version]
- Inman, M.; Lovato, L.; Doster, A.; Jones, C. A mutation in the latency related gene of bovine herpesvirus 1 interferes with the latency-reactivation cycle of latency in calves. J. Virol. 2002, 76, 6771–6779. [Google Scholar] [CrossRef] [Green Version]
- Lovato, L.; Inman, M.; Henderson, G.; Doster, A.; Jones, C. Infection of cattle with a bovine herpesvirus 1 (BHV-1) strain that contains a mutation in the latency related gene leads to increased apoptosis in trigeminal ganglia during the transition from acute infection to latency. J. Virol. 2003, 77, 4848–4857. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Jones, C. Open reading frame 2, encoded by the latency-related gene of bovine herpesvirus 1, has antiapoptotic activity in transiently transfected neuroblastoma cells. J. Virol. 2008, 82, 10940–10945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornell, R.A.; Eisen, J.S. Notch in the pathway: The roles of Notch signaling in neural crest development. Semin. Cell Dev. Biol. 2005, 16, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Workman, A.; Sinani, D.; Pittayakhajonwut, D.; Jones, C. A protein (ORF2) encoded by the latency related gene of bovine herpesvirus 1 interacts with Notch1 and Notch3. J. Virol. 2011, 85, 2536–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Jones, C. Regulation of Notch-mediated transcription by a bovine herpesvirus 1 encoded protein (ORF2) that is expressed in latently infected sensory neurons. J. Neurovirol. 2016, 22, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Sinani, D.; Jones, C. Localization of sequences in a protein (ORF2) encoded by the latency-related gene of bovine herpesvirus 1 that inhibits apoptosis and interferes with Notch1-mediated trans-activation of the bICP0 promoter. J. Virol. 2011, 85, 12124–12133. [Google Scholar] [CrossRef] [Green Version]
- Decman, V.; Freeman, M.L.; Kinchington, P.R.; Hendricks, R.L. Immune control of HSV-1 latency. Viral. Immunol. 2005, 18, 466–473. [Google Scholar] [CrossRef]
- Khanna, K.M.; Lepisto, A.J.; Decman, V.; Hendricks, R.L. Immune control of herpes simplex virus during latency. Curr. Opin. Immunol. 2004, 16, 463–469. [Google Scholar] [CrossRef]
- Hendricks, R.L.; Weber, P.C.; Taylor, J.L.; Koumbis, A.; Tumpey, T.M.; Glorioso, J.C. Endogenously produced interferon alpha protects mice from herpes simplex virus type 1 corneal disease. J. Gen. Virol. 1991, 72, 1601–1610. [Google Scholar] [CrossRef]
- Mikloska, Z.; Cunningham, A.L. Alpha and gamma interferons inhibit herpes simplex virus type 1 infection and spread in epidermal cells after axonal transmission. J. Virol. 2001, 75, 11821–11826. [Google Scholar] [CrossRef] [Green Version]
- Sainz, B., Jr.; Halford, W.P. Alpha/Beta interferon and gamma interferon synergize to inhibit the replication of herpes simplex virus type 1. J. Virol. 2002, 76, 11541–11550. [Google Scholar] [CrossRef] [Green Version]
- De Regge, N.; Van Opdenbosch, N.; Nauwynck, H.J.; Efstathiou, S.; Favoreel, H.W. Interferon alpha induces establishment of alphaherpesvirus latency in sensory neurons in vitro. PLoS ONE 2010, 5, e13076. [Google Scholar] [CrossRef] [PubMed]
- Maroui, M.A.; Callé, A.; Cohen, C.; Streichenberger, N.; Texier, P.; Takissian, J.; Rousseau, A.; Poccardi, N.; Welsch, J.; Corpet, A.; et al. Latency entry of herpes simplex virus 1 is determined by the interaction of its genome with the nuclear environment. PLoS Pathog. 2016, 12, e1005834. [Google Scholar] [CrossRef] [PubMed]
- Tormanen, K.; Allen, S.; Mott, K.R.; Ghiasi, H. The latency-associated transcript inhibits apoptosis via downregulation of components of the type I interferon pathway during latent herpes simplex virus 1 ocular infection. J. Virol. 2019, 93, e00103-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Tang, J. Evasion of I interferon-mediated innate immunity by pseudorabies virus. Front. Microbiol. 2021, 12, 801257. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Cao, M.; Bai, J.; Jin, L.; Wang, X.; Gao, Y.; Liu, X.; Jiang, P. PRV-encoded UL13 protein kinase acts as an antagonist of innate immunity by targeting IRF3-signaling pathways. Vet. Microbiol. 2020, 250, 108860. [Google Scholar] [CrossRef]
- Zhang, R.; Chen, S.; Zhang, Y.; Wang, M.; Qin, C.; Yu, C.; Zhang, Y.; Li, Y.; Chen, L.; Zhang, X.; et al. Pseudorabies virus DNA polymerase processivity factor UL42 inhibits type I IFN response by preventing ISGF3-ISRE interaction. J. Immunol. 2021, 207, 613–625. [Google Scholar] [CrossRef]
- Zhang, R.; Xu, A.; Qin, C.; Zhang, Q.; Chen, S.; Lang, Y.; Wang, M.; Li, C.; Feng, W.; Zhang, R.; et al. Pseudorabies virus dUTPase UL50 induces lysosomal degradation of type I interferon receptor 1 and antagonizes the alpha interferon response. J. Virol. 2017, 91, e01148-17. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Kong, N.; Xu, J.; Wang, J.; Zhang, M.; Ruan, K.; Li, L.; Zhang, Y.; Zheng, H.; Tong, W.; et al. Pseudorabies virus UL24 antagonizes OASL-mediated antiviral effect. Virus Res. 2021, 295, 198276. [Google Scholar] [CrossRef]
- Camarena, V.; Kobayashi, M.; Kim, J.Y.; Roehm, P.; Perez, R.; Gardner, J.; Wilson, A.C.; Mohr, I.; Chao, M.V. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 2010, 8, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Koyuncu, O.O.; MacGibeny, M.A.; Hogue, I.B.; Enquist, L.W. Compartmented neuronal cultures reveal two distinct mechanisms for alpha herpesvirus escape from genome silencing. PLoS Pathog. 2017, 13, e1006608. [Google Scholar] [CrossRef] [Green Version]
- Sciammas, R.; Kodukula, P.; Tang, Q.; Hendricks, R.L.; Bluestone, J.A. T cell receptor-gamma/delta cells protect mice from herpes simplex virus type 1-induced lethal encephalitis. J. Exp. Med. 1997, 185, 1969–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodukula, P.; Liu, T.; Rooijen, N.V.; Jager, M.J.; Hendricks, R.L. Macrophage control of herpes simplex virus type 1 replication in the peripheral nervous system. J. Immunol. 1999, 162, 2895–2905. [Google Scholar] [PubMed]
- Marrack, P.; Kappler, J. The T cell receptor. Science 1987, 238, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Simmons, A. H-2-linked genes influence the severity of herpes simplex virus infection of the peripheral nervous system. J. Exp. Med. 1989, 169, 1503–1507. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.; Rowe, A.; Hendricks, R. Increased pathology in herpes stromal keratitis results from differences in local, and not systemic, immune responses (VIR5P. 1028). J. Immunol. 2014, 192 (Suppl. 1), 144.11. [Google Scholar]
- Liu, T.; Khanna, K.M.; Chen, X.; Fink, D.J.; Hendricks, R.L. CD8+ T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J. Exp. Med. 2000, 191, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Simmons, A.; Tscharke, D.; Speck, P. The role of immune mechanisms in control of herpes simplex virus infection of the peripheral nervous system. Curr. Top. Microbiol. Immunol. 1992, 179, 31–56. [Google Scholar]
- Shimeld, C.; Whiteland, J.L.; Williams, N.A.; Easty, D.L.; Hill, T.J. Cytokine production in the nervous system of mice during acute and latent infection with herpes simplex virus type 1. J. Gen. Virol. 1997, 78, 3317–3325. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Khanna, K.M.; Carriere, B.N.; Hendricks, R.L. Gamma interferon can prevent herpes simplex virus type 1 reactivation from latency in sensory neurons. J. Virol. 2001, 75, 11178–11184. [Google Scholar] [CrossRef] [Green Version]
- Cantin, E.; Tanamachi, B.; Openshaw, H. Role for gamma interferon in control of herpes simplex virus type 1 reactivation. J. Virol. 1999, 73, 3418–3423. [Google Scholar] [CrossRef] [Green Version]
- Davido, D.J.; Leib, D.A.; Schaffer, P.A. The cyclin-dependent kinase inhibitor roscovitine inhibits the transactivating activity and alters the posttranslational modification of herpes simplex virus type 1 ICP0. J. Virol. 2002, 76, 1077–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, M.; Bandyopadhyay, D.; Goepfert, T.M.; Kumar, R. Interferon induces expression of cyclin-dependent kinase-inhibitors p21WAF1 and p27Kip1 that prevent activation of cyclindependent kinase by CDK-activating kinase (CAK). Oncogene 1998, 16, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treat, B.R.; Bidula, S.M.; St Leger, A.J.; Hendricks, R.L.; Kinchington, P.R. Herpes simplex virus 1-specific CD8+ T cell priming and latent ganglionic retention are shaped by viral epitope promoter kinetics. J. Virol. 2020, 94, e01193-19. [Google Scholar] [CrossRef]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Pesnicak, L.; Cohen, J.I.; Straus, S.E. Rates of reactivation of latent herpes simplex virus from mouse trigeminal ganglia ex vivo correlate directly with viral load and inversely with number of infiltrating CD8+ T cells. J. Virol. 2007, 81, 8157–8164. [Google Scholar] [CrossRef] [Green Version]
- Chentoufi, A.A.; Dasgupta, G.; Christensen, N.D.; Hu, J.; Choudhury, Z.S.; Azeem, A.; Jester, J.V.; Nesburn, A.B.; Wechsler, S.L.; BenMohamed, L. A novel HLA (HLA-A*0201) transgenic rabbit model for preclinical evaluation of human CD8+ T cell epitopebased vaccines against ocular herpes. J. Immunol. 2010, 184, 2561–2571. [Google Scholar] [CrossRef] [Green Version]
- Coulon, P.G.; Roy, S.; Prakash, S.; Srivastava, R.; Dhanushkodi, N.; Salazar, S.; Amezquita, C.; Nguyen, L.; Vahed, H.; Nguyen, A.M.; et al. Upregulation of multiple CD8+ T cell exhaustion pathways is associated with recurrent ocular herpes simplex virus type 1 infection. J. Immunol. 2020, 205, 454–468. [Google Scholar] [CrossRef]
- Frank, G.M.; Lepisto, A.J.; Freeman, M.L.; Sheridan, B.S.; Cherpes, T.L.; Hendricks, R.L. Early CD4+ T cell help prevents partial CD8+ T cell exhaustion and promotes maintenance of herpes simplex virus 1 latency. J. Immunol. 2010, 184, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Geng, S.; Suo, Y.; Wei, X.; Cai, Q.; Wu, B.; Zhou, X.; Shi, Y.; Wang, B. Critical role of regulatory T cells in the latency and stress-induced reactivation of HSV-1. Cell Rep. 2018, 25, 2379–2389.e3. [Google Scholar] [CrossRef] [Green Version]
- Mengeling, W.L.; Lager, K.M.; Volz, D.M.; Brockmeier, S.L. Effect of various vaccination procedures on shedding, latency, and reactivation of attenuated and virulent pseudorabies virus in swine. Am. J. Vet. Res. 1992, 53, 2164–2173. [Google Scholar] [PubMed]
- Vilnis, A.; Sussman, M.D.; Thacker, B.L.; Sennm, M.; Maes, R.K. Vaccine genotype and route of administration affect pseudorabies field virus latency load after challenge. Vet. Microbiol. 1998, 62, 81–96. [Google Scholar] [CrossRef]
- Schang, L.M.; Kutish, G.F.; Osorio, F.A. Correlation between precolonization of trigeminal ganglia by attenuated strains of pseudorabies virus and resistance to wild-type virus latency. J. Virol. 1994, 68, 8470–8476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, A.T.; Moonen-Leusen, H.W.; van Milligen, F.J.; Savelkoul, H.F.; Zwart, R.J.; Kimman, T.G. A mouse model to study immunity against pseudorabies virus infection: Significance of CD4+ and CD8+ cells in protective immunity. Vaccine 1998, 16, 1550–1558. [Google Scholar] [CrossRef]
- Knipe, D.M.; Lieberman, P.M.; Jung, J.U.; McBride, A.A.; Morris, K.V.; Ott, M.; Margolis, D.; Nieto, A.; Nevels, M.; Parks, R.J.; et al. Snapshots: Chromatin control of viral infection. Virology 2013, 435, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Rock, D.L.; Fraser, N.W. Detection of HSV-1 genome in central nervous system of latently infected mice. Nature 1983, 302, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Deshmane, S.L.; Fraser, N.W. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J. Virol. 1989, 63, 943–947. [Google Scholar] [CrossRef] [Green Version]
- Wang, O.Y.; Zhou, C.; Johnson, K.E.; Colgrove, R.C.; Coen, D.M.; Knipe, D.M. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 2005, 102, 16055–16059. [Google Scholar] [CrossRef] [Green Version]
- Cliffe, A.R.; Garber, D.A.; Knipe, D.M. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 2009, 83, 8182–8190. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, D.L.; Thompson, H.W.; Bloom, D.C. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J. Virol. 2009, 83, 8173–8181. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.H.; Ponce de Leon, M.; Diggelmann, H.; Lawrence, W.C.; Vernon, S.K.; Eisenberg, R.J. Structural analysis of the capsid polypeptides of herpes simplex virus types 1 and 2. J. Virol. 1980, 34, 521–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignatti, P.F.; Cassai, E. Analysis of herpes simplex virus nucleoprotein complexes extracted from infected cells. J. Virol. 1980, 36, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Cereghini, S.; Yaniv, M. Assembly of transfected DNA into chromatin: Structural changes in the origin-promoter-enhancer region upon replication. EMBO J. 1984, 3, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Monier, K.; Armas, J.C.; Etteldorf, S.; Ghazal, P.; Sullivan, K.F. Annexation of the interchromosomal space during viral infection. Nature Cell Biol. 2000, 2, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Simpson-Holley, M.; Baines, J.; Roller, R.; Knipe, D.M. Herpes simplex virus 1 UL31 and UL34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J. Virol. 2004, 78, 5591–5600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, F.J.; Triezenberg, S.J. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J. Virol. 2004, 78, 9689–9696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Knipe, D.M. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 2008, 82, 12030–12038. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.J.; Han, S.S.; Shi, Q.H.; Shi, A.; Yang, L.; Chang, H.T.; Zhao, J.; Wang, C.Q.; Chen, L. Chromatin state of some genome segment of pseudorabies virus during lytic infection. Chin. J. Vet. Sci. 2017, 37, 585–591. (In Chinese) [Google Scholar]
- Workman, J.L.; Abmayr, S.M.; Cromlish, W.A.; Roeder, R.G. Transcriptional regulation by the immediate early protein of pseudorabies virus during in vitro nucleosome assembly. Cell 1988, 55, 211–219. [Google Scholar] [CrossRef]
- Koyuncu, O.O.; Song, R.; Greco, T.M.; Cristea, I.M.; Enquist, L.W. The number of alphaherpesvirus particles infecting axons and the axonal protein repertoire determines the outcome of neuronal infection. MBio 2015, 6, e00276-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Viral Genes | Viral Proteins | Virus | Proposed Function |
|---|---|---|---|
| UL23 | Thymidine kinase (TK) | HSV-1 | Necessary for HSV-1 reactivation and unnecessary for latency establishment |
| HSV-2 | Important but nonessential for latency or reactivation | ||
| BHV-1 | Probably unnecessary for latency-reactivation cycle | ||
| PRV | Crucial for the latent PRV reactivation | ||
| US8 | gE | HSV-1/PRV/BHV-1 | Important for efficient virus latency establishment and reactivation in neurons due to its capacity for promoting virus neuroinvasion |
| US9 | 11K | HSV-1/PRV | Affect virus reactivation due to its critical role in promoting virus anterograde transport in neurons |
| EP0 | EP0 | HSV-1/PRV | Crucial for the latency and reactivation |
| IE180 | IE180 | PRV | Has important roles in PRV reactivation; Important for swiching from latency to reactivation state |
| ORF1 | ORF1 | BHV-1 | Might be important for the virus latency and reactivation |
| ORF2 | ORF2 | BHV-1 | Inhibits apoptosis; interferes with Notch1-mediated transactivation of ICP0; probably inhibit virus productive infection and promote virus latency |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Li, G.; Wan, C.; Li, Y.; Peng, L.; Fang, R.; Peng, Y.; Ye, C. A Comparison of Pseudorabies Virus Latency to Other α-Herpesvirinae Subfamily Members. Viruses 2022, 14, 1386. https://doi.org/10.3390/v14071386
Chen J, Li G, Wan C, Li Y, Peng L, Fang R, Peng Y, Ye C. A Comparison of Pseudorabies Virus Latency to Other α-Herpesvirinae Subfamily Members. Viruses. 2022; 14(7):1386. https://doi.org/10.3390/v14071386
Chicago/Turabian StyleChen, Jing, Gang Li, Chao Wan, Yixuan Li, Lianci Peng, Rendong Fang, Yuanyi Peng, and Chao Ye. 2022. "A Comparison of Pseudorabies Virus Latency to Other α-Herpesvirinae Subfamily Members" Viruses 14, no. 7: 1386. https://doi.org/10.3390/v14071386