
Origin and Deep Evolution of Human Endogenous Retroviruses in Pan-Primates


Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Screening and Identification of HERVs
2.2. Vertical Transmission Identification
2.3. Genomic Rearrangement Analysis
2.4. HERVs-Derived ncRNA Verification in the Human Genome
3. Results
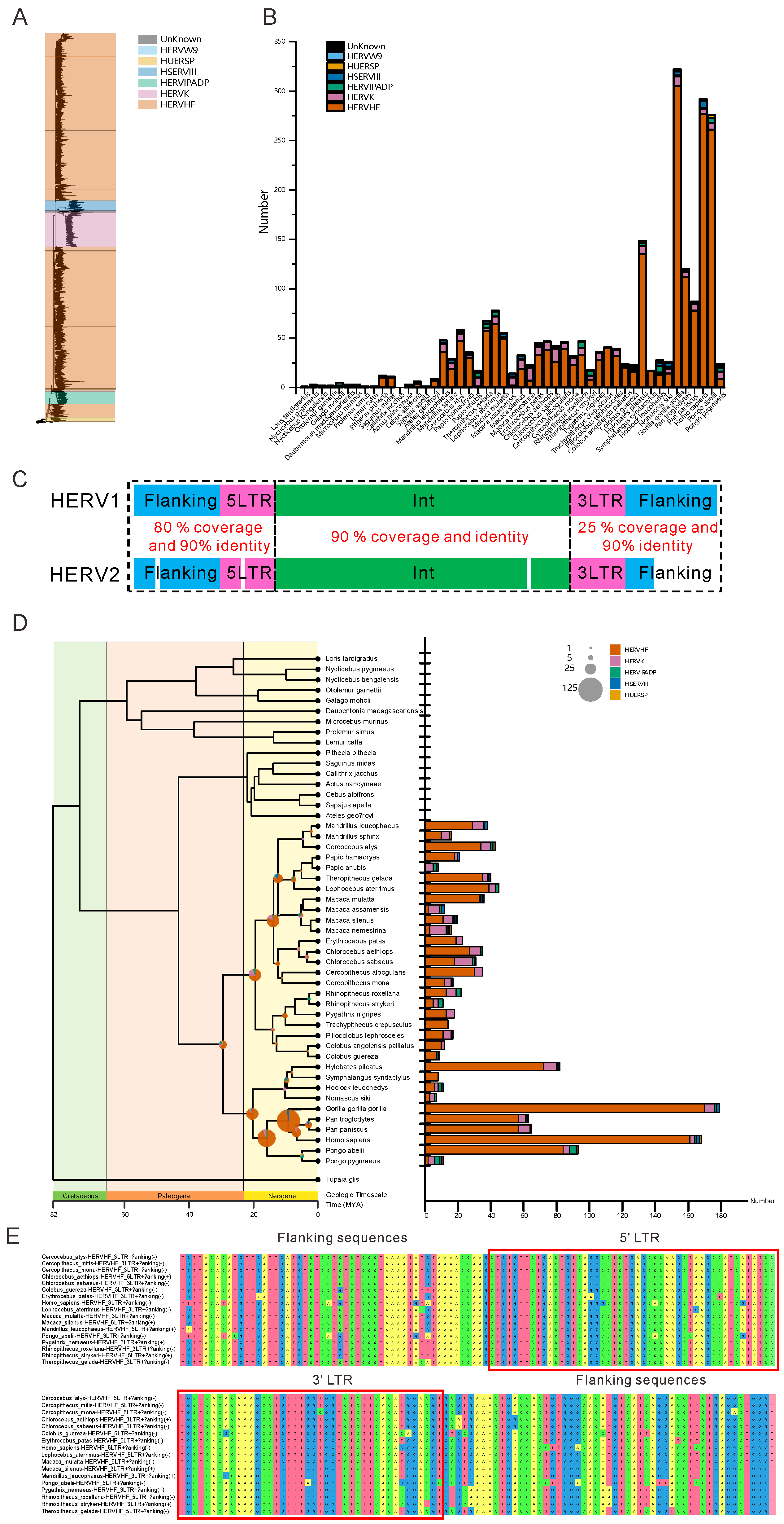
3.1. HERVs Are Widely Dispersed in the Genomes of Old World Monkeys and Apes
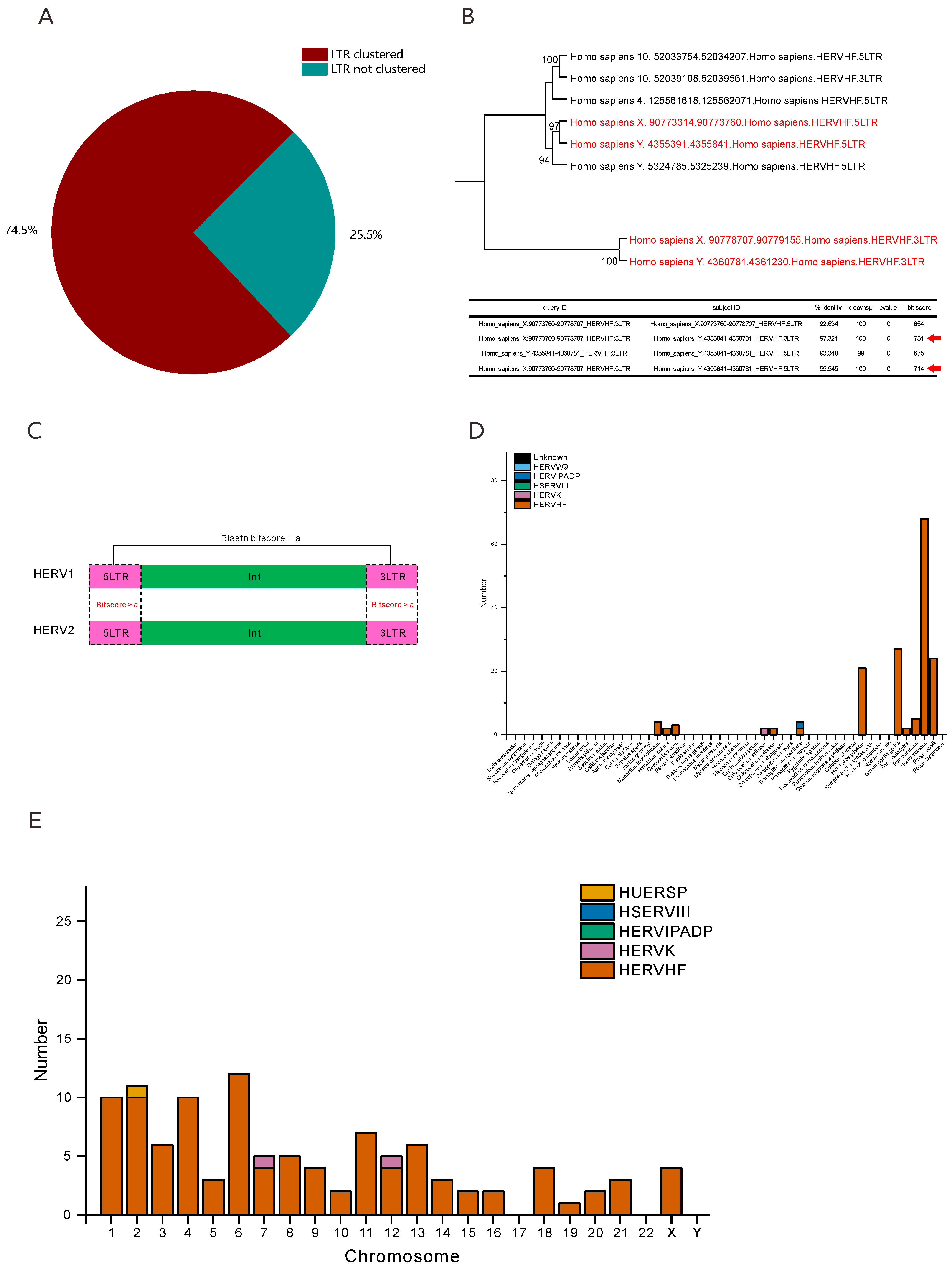
3.2. Numerous HERVHFs Are Spread by Vertical Transmission within Catarrhini
3.3. Some F-HERVs May Be Involved in Genomic Rearrangement
3.4. Some F-HERVs in Human Genomes Are Likely Transcribed into ncRNAs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, W.E. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat. Rev. Microbiol. 2019, 17, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, D.J. Endogenous retroviruses in the human genome sequence. Genome Biol. 2001, 2, reviews1017.1. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, A.D.; Ishida, Y.; O’Brien, S.P.; Roca, A.L.; Eiden, M.V. Transmission, Evolution, and Endogenization: Lessons Learned from Recent Retroviral Invasions. Microbiol. Mol. Biol. Rev. 2018, 82, e00044-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Montojo, M.; Doucet-O’Hare, T.; Henderson, L.; Nath, A. Human endogenous retrovirus-K (HML-2): A comprehensive review. Crit. Rev. Microbiol. 2018, 44, 715–738. [Google Scholar] [CrossRef] [PubMed]
- Jansz, N.; Faulkner, G.J. Endogenous retroviruses in the origins and treatment of cancer. Genome Biol. 2021, 22, 147. [Google Scholar] [CrossRef]
- Martin, M.A.; Bryan, T.; Rasheed, S.; Khan, A.S. Identification and cloning of endogenous retroviral sequences present in human DNA. Proc. Natl. Acad. Sci. USA 1981, 78, 4892–4896. [Google Scholar] [CrossRef] [Green Version]
- Hayward, A.; Cornwallis, C.K.; Jern, P. Pan-vertebrate comparative genomics unmasks retrovirus macroevolution. Proc. Natl. Acad. Sci. USA 2015, 112, 464–469. [Google Scholar] [CrossRef] [Green Version]
- Vargiu, L.; Rodriguez-Tome, P.; Sperber, G.O.; Cadeddu, M.; Grandi, N.; Blikstad, V.; Tramontano, E.; Blomberg, J. Classification and characterization of human endogenous retroviruses; mosaic forms are common. Retrovirology 2016, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Bannert, N.; Kurth, R. The evolutionary dynamics of human endogenous retroviral families. Annu. Rev. Genom. Hum. Genet. 2006, 7, 149–173. [Google Scholar] [CrossRef]
- Escalera-Zamudio, M.; Greenwood, A.D. On the classification and evolution of endogenous retrovirus: Human endogenous retroviruses may not be ‘human’ after all. APMIS 2016, 124, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Mager, D.L.; Stoye, J.P. Mammalian Endogenous Retroviruses. Microbiol. Spectr. 2015, 3, MDNA3-0009-2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tristem, M. Identification and characterization of novel human endogenous retrovirus families by phylogenetic screening of the human genome mapping project database. J. Virol. 2000, 74, 3715–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, S.; Takahashi, M.U. gEVE: A genome-based endogenous viral element database provides comprehensive viral protein-coding sequences in mammalian genomes. Database 2016, 2016, baw087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhao, H.; Gong, Z.; Han, G.Z. Endogenous retroviruses of non-avian/mammalian vertebrates illuminate diversity and deep history of retroviruses. PLoS Pathog. 2018, 14, e1007072. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Yu, G. Scatterpie: Scatter Pie Plot. 2021. Available online: https://guangchuangyu.github.io/scatterpie/ (accessed on 1 December 2016).
- Hughes, J.F.; Coffin, J.M. Evidence for genomic rearrangements mediated by human endogenous retroviruses during primate evolution. Nat. Genet. 2001, 29, 487–489. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, J.; Li, Y.; Song, T.; Wu, Y.; Fang, S.; Bu, D.; Li, H.; Sun, L.; Pei, D.; et al. NONCODEV6: An updated database dedicated to long non-coding RNA annotation in both animals and plants. Nucleic Acids Res. 2021, 49, D165–D171. [Google Scholar] [CrossRef]
- Xiong, Y.; Eickbush, T.H. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 1990, 9, 3353–3362. [Google Scholar] [CrossRef]
- De Parseval, N.; Heidmann, T. Human endogenous retroviruses: From infectious elements to human genes. Cytogenet. Genome Res. 2005, 110, 318–332. [Google Scholar] [CrossRef]
- Mager, D.L.; Freeman, J.D. HERV-H endogenous retroviruses: Presence in the New World branch but amplification in the Old World primate lineage. Virology 1995, 213, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Sverdlov, E.D. Retroviruses and primate evolution. Bioessays 2000, 22, 161–171. [Google Scholar] [CrossRef]
- Yi, J.M.; Kim, H.S. Evolutionary implication of human endogenous retrovirus HERV-H family. J. Hum. Genet. 2004, 49, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodchild, N.L.; Wilkinson, D.A.; Mager, D.L. Recent evolutionary expansion of a subfamily of RTVL-H human endogenous retrovirus-like elements. Virology 1993, 196, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Grandi, N.; Cadeddu, M.; Blomberg, J.; Mayer, J.; Tramontano, E. HERV-W group evolutionary history in non-human primates: Characterization of ERV-W orthologs in Catarrhini and related ERV groups in Platyrrhini. BMC Evol. Biol. 2018, 18, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloway, J.R.; Williams, Z.H.; Freeman, M.M.; Bulow, U.; Coffin, J.M. Gorillas have been infected with the HERV-K (HML-2) endogenous retrovirus much more recently than humans and chimpanzees. Proc. Natl. Acad. Sci. USA 2019, 116, 1337–1346. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Kim, H.S. Endogenous retrovirus HERV-I LTR family in primates: Sequences, phylogeny, and evolution. Arch. Virol. 2006, 151, 1651–1658. [Google Scholar] [CrossRef]
- Yi, J.M.; Kim, T.H.; Huh, J.W.; Park, K.S.; Jang, S.B.; Kim, H.M.; Kim, H.S. Human endogenous retroviral elements belonging to the HERV-S family from human tissues, cancer cells, and primates: Expression, structure, phylogeny and evolution. Gene 2004, 342, 283–292. [Google Scholar] [CrossRef]
- Shah, A.H.; Gilbert, M.; Ivan, M.E.; Komotar, R.J.; Heiss, J.; Nath, A. The role of human endogenous retroviruses in gliomas: From etiological perspectives and therapeutic implications. Neuro Oncol. 2021, 23, 1647–1655. [Google Scholar] [CrossRef]
- Srinivasachar Badarinarayan, S.; Sauter, D. Switching Sides: How Endogenous Retroviruses Protect Us from Viral Infections. J. Virol. 2021, 95, e02299-20. [Google Scholar] [CrossRef]
- Xiang, Y.; Liang, H. The Regulation and Functions of Endogenous Retrovirus in Embryo Development and Stem Cell Differentiation. Stem Cells Int. 2021, 2021, 6660936. [Google Scholar] [CrossRef]
- Campbell, I.M.; Gambin, T.; Dittwald, P.; Beck, C.R.; Shuvarikov, A.; Hixson, P.; Patel, A.; Gambin, A.; Shaw, C.A.; Rosenfeld, J.A.; et al. Human endogenous retroviral elements promote genome instability via non-allelic homologous recombination. BMC Biol. 2014, 12, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trombetta, B.; Fantini, G.; D’Atanasio, E.; Sellitto, D.; Cruciani, F. Evidence of extensive non-allelic gene conversion among LTR elements in the human genome. Sci. Rep. 2016, 6, 28710. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.D.; Ameen, M.; Guo, H.; Abilez, O.J.; Tian, L.; Mumbach, M.R.; Diecke, S.; Qin, X.; Liu, Y.; Yang, H.; et al. Endogenous Retrovirus-Derived lncRNA BANCR Promotes Cardiomyocyte Migration in Humans and Non-human Primates. Dev. Cell 2020, 54, 694–709.e699. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Qi, F.; Wu, F.; Nie, H.; Song, Y.; Shao, L.; Han, J.; Wu, Z.; Saiyin, H.; Wei, G.; et al. Endogenous Retrovirus-Derived Long Noncoding RNA Enhances Innate Immune Responses via Derepressing RELA Expression. MBio 2019, 10, e00937-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Pi, W.; Zhu, X.; Yu, M.; Ha, H.; Shi, H.; Choi, J.H.; Tuan, D. Long non-coding RNAs transcribed by ERV-9 LTR retrotransposon act in cis to modulate long-range LTR enhancer function. Nucleic Acids Res. 2017, 45, 4479–4492. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.K. AcademH, a lineage of Academ DNA transposons encoding helicase found in animals and fungi. Mob. DNA 2020, 11, 15. [Google Scholar] [CrossRef]
- Xiong, Y.; Eickbush, T.H. Similarity of reverse transcriptase-like sequences of viruses, transposable elements, and mitochondrial introns. Mol. Biol. Evol. 1988, 5, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Nolan, A.; Watson, J.; Tristem, M. Identification of an ancient endogenous retrovirus, predating the divergence of the placental mammals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120503. [Google Scholar] [CrossRef] [Green Version]
- Jern, P.; Sperber, G.O.; Blomberg, J. Divergent patterns of recent retroviral integrations in the human and chimpanzee genomes: Probable transmissions between other primates and chimpanzees. J. Virol. 2006, 80, 1367–1375. [Google Scholar] [CrossRef] [Green Version]
- Magiorkinis, G.; Blanco-Melo, D.; Belshaw, R. The decline of human endogenous retroviruses: Extinction and survival. Retrovirology 2015, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Bosch, E.; Jobling, M.A. Duplications of the AZFa region of the human Y chromosome are mediated by homologous recombination between HERVs and are compatible with male fertility. Hum. Mol. Genet. 2003, 12, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, C.; Voet, T.; Zamani Esteki, M.; Nowakowska, B.A.; Vermeesch, J.R. Nonallelic homologous recombination between retrotransposable elements is a driver of de novo unbalanced translocations. Genome Res. 2013, 23, 411–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weckselblatt, B.; Hermetz, K.E.; Rudd, M.K. Unbalanced translocations arise from diverse mutational mechanisms including chromothripsis. Genome Res. 2015, 25, 937–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löber, U.; Hobbs, M.; Dayaram, A.; Tsangaras, K.; Jones, K.; Alquezar-Planas, D.E.; Ishida, Y.; Meers, J.; Mayer, J.; Quedenau, C.; et al. Degradation and remobilization of endogenous retroviruses by recombination during the earliest stages of a germ-line invasion. Proc. Natl. Acad. Sci. USA 2018, 115, 8609–8614. [Google Scholar] [CrossRef] [Green Version]
- Ariza, M.E.; Williams, M.V. A human endogenous retrovirus K dUTPase triggers a TH1, TH17 cytokine response: Does it have a role in psoriasis? J. Investig. Dermatol. 2011, 131, 2419–2427. [Google Scholar] [CrossRef] [Green Version]
- Volkman, H.E.; Stetson, D.B. The enemy within: Endogenous retroelements and autoimmune disease. Nat. Immunol. 2014, 15, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Srinivasachar Badarinarayan, S.; Shcherbakova, I.; Langer, S.; Koepke, L.; Preising, A.; Hotter, D.; Kirchhoff, F.; Sparrer, K.M.J.; Schotta, G.; Sauter, D. HIV-1 infection activates endogenous retroviral promoters regulating antiviral gene expression. Nucleic Acids Res. 2020, 48, 10890–10908. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Chromosome | Start | End | HERVname | Strand | Related-ncRNA |
|---|---|---|---|---|---|
| 1 | 22997488 | 23002547 | Homo_sapiens_1_23000272-23002212-HERVHF | − | NONHSAG057580.1 |
| 1 | 43087974 | 43091095 | Homo_sapiens_1_43087972-43089561-HERVHF | + | NONHSAG056389.1 |
| 1 | 68386003 | 68391994 | Homo_sapiens_1_68388791-68390135-HERVHF | + | NONHSAG001773.3 |
| 1 | 82354581 | 82360561 | Homo_sapiens_1_82356924-82357985-HERVHF | − | ENSG00000233290 |
| 1 | 82955299 | 82961592 | Homo_sapiens_1_82956772-82959208-HERVHF | + | ENSG00000230817 |
| 1 | 209451675 | 209454012 | Homo_sapiens_1_209451456-209452546-HERVHF | + | NONHSAG057153.1 |
| 1 | 224840009 | 224846095 | Homo_sapiens_1_224842067-224843662-HERVHF | − | ENSG00000286719 |
| 1 | 228942542 | 228942868 | Homo_sapiens_1_228948757-228950104-HERVHF | + | NONHSAG057288.1 |
| 1 | 232120241 | 232123943 | Homo_sapiens_1_232120704-232121847-HERVHF | + | NONHSAG004630.2 |
| 1 | 241433890 | 241439885 | Homo_sapiens_1_241436435-241438237-HERVHF | + | ENSG00000287516 |
| 2 | 5000768 | 5003355 | Homo_sapiens_2_5003505-5005037-HERVHF | + | NONHSAG077068.1 |
| 2 | 16791950 | 16797713 | Homo_sapiens_2_16793801-16795145-HERVHF | − | NONHSAG078497.1 |
| 2 | 34789818 | 34796058 | Homo_sapiens_2_34792231-34793755-HERVHF | + | NONHSAG077257.1 |
| 2 | 38080800 | 38086513 | Homo_sapiens_2_38082627-38083973-HERVHF | − | ENSG00000138061 |
| 2 | 67333734 | 67337603 | Homo_sapiens_2_67334137-67335887-HERVHF | + | NONHSAG028042.3 |
| 2 | 69789900 | 69795859 | Homo_sapiens_2_69792965-69796021-HUERSP | − | NONHSAG028084.2 |
| 2 | 77965137 | 77970868 | Homo_sapiens_2_77967627-77968976-HERVHF | + | NONHSAG077496.1 |
| 2 | 192506078 | 192513184 | Homo_sapiens_2_192509946-192511342-HERVHF | + | NONHSAG030125.2 |
| 2 | 215922303 | 215928129 | Homo_sapiens_2_215924899-215926434-HERVHF | + | NONHSAG078151.1 |
| 2 | 224225331 | 224230988 | Homo_sapiens_2_224227814-224229160-HERVHF | + | NONHSAG078214.1 |
| 2 | 237606784 | 237611630 | Homo_sapiens_2_237609479-237610820-HERVHF | + | NONHSAG110040.1 |
| 3 | 21189031 | 21194139 | Homo_sapiens_3_21190643-21192440-HERVHF | − | ENSG00000282987 |
| 3 | 54634482 | 54638068 | Homo_sapiens_3_54636349-54637755-HERVHF | − | ENSG00000265992 |
| 3 | 112418410 | 112423366 | Homo_sapiens_3_112419312-112420768-HERVHF | − | NONHSAG035734.2 |
| 3 | 115798715 | 115799166 | Homo_sapiens_3_115795176-115796709-HERVHF | − | NONHSAG085690.1 |
| 3 | 155274423 | 155278762 | Homo_sapiens_3_155276457-155278448-HERVHF | − | NONHSAG036456.2 |
| 3 | 186660747 | 186663692 | Homo_sapiens_3_186660542-186661888-HERVHF | + | ENSG00000113905 |
| 4 | 3927445 | 3930682 | Homo_sapiens_4_3929901-3931242-HERVHF | + | NONHSAG087348.2 |
| 4 | 17000545 | 17003928 | Homo_sapiens_4_17000127-17001778-HERVHF | + | NONHSAG037572.2 |
| 4 | 24500975 | 24501427 | Homo_sapiens_4_24503534-24505060-HERVHF | + | NONHSAG037630.2 |
| 4 | 27974874 | 27981319 | Homo_sapiens_4_27976550-27977552-HERVHF | + | NONHSAG037691.2 |
| 4 | 92271492 | 92275299 | Homo_sapiens_4_92273363-92274770-HERVHF | − | ENSG00000249152 |
| 4 | 103553770 | 103557353 | Homo_sapiens_4_103555460-103556971-HERVHF | − | ENSG00000250920 |
| 4 | 128640901 | 128644450 | Homo_sapiens_4_128642726-128644003-HERVHF | − | NONHSAG088517.2 |
| 4 | 145698823 | 145703505 | Homo_sapiens_4_145701612-145702617-HERVHF | + | ENSG00000237136 |
| 4 | 152741345 | 152747172 | Homo_sapiens_4_152743196-152744540-HERVHF | − | NONHSAG039129.2 |
| 4 | 175461163 | 175467003 | Homo_sapiens_4_175463047-175464647-HERVHF | − | ENSG00000249945 |
| 5 | 92826033 | 92829706 | Homo_sapiens_5_92826486-92827829-HERVHF | + | ENSG00000248588 |
| 5 | 136303790 | 136307028 | Homo_sapiens_5_136303833-136305180-HERVHF | + | ENSG00000250947 |
| 5 | 161245405 | 161254586 | Homo_sapiens_5_161251016-161252646-HERVHF | + | NONHSAG090654.1 |
| 6 | 16259010 | 16264893 | Homo_sapiens_6_16260854-16262201-HERVHF | − | ENSG00000282024 |
| 6 | 18754142 | 18756902 | Homo_sapiens_6_18755932-18757277-HERVHF | − | NONHSAG043117.2 |
| 6 | 80509795 | 80515805 | Homo_sapiens_6_80511941-80513513-HERVHF | − | NONHSAG113295.1 |
| 6 | 94553917 | 94559610 | Homo_sapiens_6_94555806-94557152-HERVHF | − | NONHSAG044390.2 |
| 6 | 97779489 | 97785327 | Homo_sapiens_6_97782122-97783636-HERVHF | + | ENSG00000271860 |
| 6 | 123582333 | 123588007 | Homo_sapiens_6_123584156-123585562-HERVHF | − | ENSG00000186439 |
| 6 | 125701846 | 125707764 | Homo_sapiens_6_125703727-125705069-HERVHF | − | ENSG00000237742 |
| 6 | 126851224 | 126854456 | Homo_sapiens_6_126851273-126852794-HERVHF | + | NONHSAG044785.3 |
| 6 | 131295347 | 131301206 | Homo_sapiens_6_131297975-131299503-HERVHF | + | NONHSAG093612.2 |
| 6 | 131338799 | 131344566 | Homo_sapiens_6_131340338-131342739-HERVHF | + | NONHSAG093612.2 |
| 6 | 131903830 | 131907420 | Homo_sapiens_6_131904209-131905555-HERVHF | + | ENSG00000236673 |
| 6 | 144923164 | 144928866 | Homo_sapiens_6_144925698-144927040-HERVHF | + | NONHSAG095837.2 |
| 7 | 26024199 | 26029809 | Homo_sapiens_7_26026061-26027405-HERVHF | − | NONHSAG047156.2 |
| 7 | 34300132 | 34300573 | Homo_sapiens_7_34301985-34303122-HERVHF | − | NONHSAG047318.2 |
| 7 | 102975230 | 102978736 | Homo_sapiens_7_102976263-102977254-HERVHF | + | ENSG00000230257 |
| 7 | 125920130 | 125924112 | Homo_sapiens_7_125920071-125921895-HERVHF | + | ENSG00000197462 |
| 7 | 155238821 | 155244070 | Homo_sapiens_7_155240740-155241657-HERVK | − | NONHSAG049243.2 |
| 8 | 71676972 | 71680514 | Homo_sapiens_8_71677433-71678968-HERVHF | + | ENSG00000254277 |
| 8 | 90090224 | 90093794 | Homo_sapiens_8_90091914-90093348-HERVHF | − | ENSG00000104327 |
| 8 | 97200769 | 97206658 | Homo_sapiens_8_97202388-97204973-HERVHF | + | NONHSAG098987.1 |
| 8 | 114284546 | 114287727 | Homo_sapiens_8_114284508-114286044-HERVHF | + | ENSG00000254339 |
| 8 | 132080235 | 132086002 | Homo_sapiens_8_132081909-132083445-HERVHF | − | ENSG00000132297 |
| 9 | 12950832 | 12954130 | Homo_sapiens_9_12950845-12952399-HERVHF | + | NONHSAG101172.2 |
| 9 | 80137297 | 80143055 | Homo_sapiens_9_80139873-80141469-HERVHF | + | NONHSAG052646.2 |
| 9 | 85461120 | 85466955 | Homo_sapiens_9_85461166-85462902-HERVHF | + | NONHSAG052703.2 |
| 9 | 115475420 | 115478923 | Homo_sapiens_9_115475976-115477349-HERVHF | + | NONHSAG053288.3 |
| 10 | 6797081 | 6802954 | Homo_sapiens_10_6798770-6800364-HERVHF | − | NONHSAG005151.3 |
| 10 | 25716420 | 25722928 | Homo_sapiens_10_25718978-25720776-HERVHF | + | ENSG00000280809 |
| 11 | 6366039 | 6371662 | Homo_sapiens_11_6368276-6369885-HERVHF | + | NONHSAG007525.2 |
| 11 | 27629072 | 27632889 | Homo_sapiens_11_27630864-27632291-HERVHF | − | ENSG00000254934 |
| 11 | 94641661 | 94647315 | Homo_sapiens_11_94644134-94645475-HERVHF | + | ENSG00000255666 |
| 11 | 96499960 | 96506627 | Homo_sapiens_11_96501724-96503654-HERVHF | + | ENSG00000183340 |
| 11 | 96590439 | 96593677 | Homo_sapiens_11_96590449-96591982-HERVHF | + | ENSG00000254587 |
| 11 | 130565609 | 130570121 | Homo_sapiens_11_130566060-130567548-HERVHF | − | NONHSAG010050.2 |
| 11 | 130753494 | 130756702 | Homo_sapiens_11_130755373-130756704-HERVHF | − | NONHSAG010050.2 |
| 12 | 4018623 | 4023691 | Homo_sapiens_12_4021109-4022208-HERVHF | + | ENSG00000256969 |
| 12 | 11462168 | 11468022 | Homo_sapiens_12_11463877-11465381-HERVHF | − | ENSG00000121335 |
| 12 | 34269097 | 34274242 | Homo_sapiens_12_34268101-34269869-HERVHF | + | NONHSAG010874.2 |
| 12 | 70444894 | 70450107 | Homo_sapiens_12_70446553-70447732-HERVK | − | NONHSAG011664.2 |
| 12 | 86941530 | 86944748 | Homo_sapiens_12_86941432-86943069-HERVHF | + | NONHSAG064903.1 |
| 13 | 42868001 | 42871007 | Homo_sapiens_13_42869513-42870545-HERVHF | − | NONHSAG013351.2 |
| 13 | 48866771 | 48872457 | Homo_sapiens_13_48868391-48870330-HERVHF | − | NONHSAG067525.1 |
| 13 | 51169866 | 51175008 | Homo_sapiens_13_51172521-51173517-HERVHF | + | NONHSAG013541.3 |
| 13 | 54127417 | 54133159 | Homo_sapiens_13_54129305-54130960-HERVHF | − | ENSG00000234787 |
| 13 | 66142250 | 66147037 | Homo_sapiens_13_66143157-66144503-HERVHF | − | NONHSAG013698.2 |
| 13 | 79276611 | 79279830 | Homo_sapiens_13_79276654-79278001-HERVHF | + | NONHSAG067153.1 |
| 14 | 38193319 | 38196529 | Homo_sapiens_14_38193317-38194783-HERVHF | + | ENSG00000258649 |
| 14 | 41521426 | 41521883 | Homo_sapiens_14_41518469-41520184-HERVHF | + | NONHSAG014802.2 |
| 14 | 48262389 | 48263146 | Homo_sapiens_14_48256895-48258584-HERVHF | − | ENSG00000287492 |
| 15 | 74354141 | 74359786 | Homo_sapiens_15_74355867-74357936-HERVHF | + | ENSG00000260266 |
| 15 | 87831107 | 87837024 | Homo_sapiens_15_87833731-87835137-HERVHF | + | NONHSAG017784.2 |
| 16 | 60078536 | 60084582 | Homo_sapiens_16_60081354-60082700-HERVHF | + | NONHSAG071739.1 |
| 16 | 65229803 | 65233421 | Homo_sapiens_16_65231504-65233039-HERVHF | − | ENSG00000260834 |
| 18 | 28693028 | 28696068 | Homo_sapiens_18_28694974-28696314-HERVHF | − | NONHSAG075074.1 |
| 18 | 56417745 | 56421344 | Homo_sapiens_18_56418118-56419466-HERVHF | + | NONHSAG074828.1 |
| 18 | 57064647 | 57070296 | Homo_sapiens_18_57068491-57069834-HERVHF | − | ENSG00000258609 |
| 18 | 73327171 | 73330369 | Homo_sapiens_18_73327166-73328509-HERVHF | + | ENSG00000261780 |
| 19 | 22568269 | 22575022 | Homo_sapiens_19_22570768-22572352-HERVHF | + | NONHSAG025320.2 |
| 20 | 12756027 | 12759632 | Homo_sapiens_20_12756400-12757916-HERVHF | + | NONHSAG031288.2 |
| 20 | 40269047 | 40274769 | Homo_sapiens_20_40271576-40272881-HERVHF | + | NONHSAG081519.1 |
| 21 | 17124024 | 17127764 | Homo_sapiens_21_17123959-17125734-HERVHF | + | NONHSAG110806.1 |
| 21 | 26227947 | 26233485 | Homo_sapiens_21_26229594-26231104-HERVHF | − | NONHSAG032575.2 |
| 21 | 42800845 | 42803999 | Homo_sapiens_21_42802518-42804296-HERVHF | − | NONHSAG083198.1 |
| X | 71264372 | 71272628 | Homo_sapiens_X_71266645-71268493-HERVHF | + | ENSG00000147140 |
| X | 94698818 | 94701832 | Homo_sapiens_X_94700559-94702091-HERVHF | − | NONHSAG054922.2 |
| X | 111543806 | 111549675 | Homo_sapiens_X_111546380-111547978-HERVHF | + | NONHSAG055109.3 |
| X | 122227333 | 122227787 | Homo_sapiens_X_122224556-122226109-HERVHF | + | NONHSAG055239.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Zhang, G.; Cui, J. Origin and Deep Evolution of Human Endogenous Retroviruses in Pan-Primates. Viruses 2022, 14, 1370. https://doi.org/10.3390/v14071370
Li Y, Zhang G, Cui J. Origin and Deep Evolution of Human Endogenous Retroviruses in Pan-Primates. Viruses. 2022; 14(7):1370. https://doi.org/10.3390/v14071370
Chicago/Turabian StyleLi, Yian, Guojie Zhang, and Jie Cui. 2022. "Origin and Deep Evolution of Human Endogenous Retroviruses in Pan-Primates" Viruses 14, no. 7: 1370. https://doi.org/10.3390/v14071370