
The Isolation and Full-Length Transcriptome Sequencing of a Novel Nidovirus and Response of Its Infection in Japanese Flounder (Paralichthys olivaceus)

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fish Sampling and Virus Isolation
2.2. Virus Genome Sequencing
2.3. Phylogenetic Analysis
2.4. RT-PCR Detection of Virus
2.5. Histology and Transmission Electron Microscopy (TEM)
2.6. Iso-Seq Library Preparation, Sequencing, Assembly, and Annotation
2.7. Isoform Structure Analysis
2.8. Transcript Factor and LncRNA Analysis
2.9. RNA-Seq by Illumina Hiseq XTen
2.10. Differential Expression Analysis and Functional Enrichment
2.11. qRT-PCR Validation
3. Results
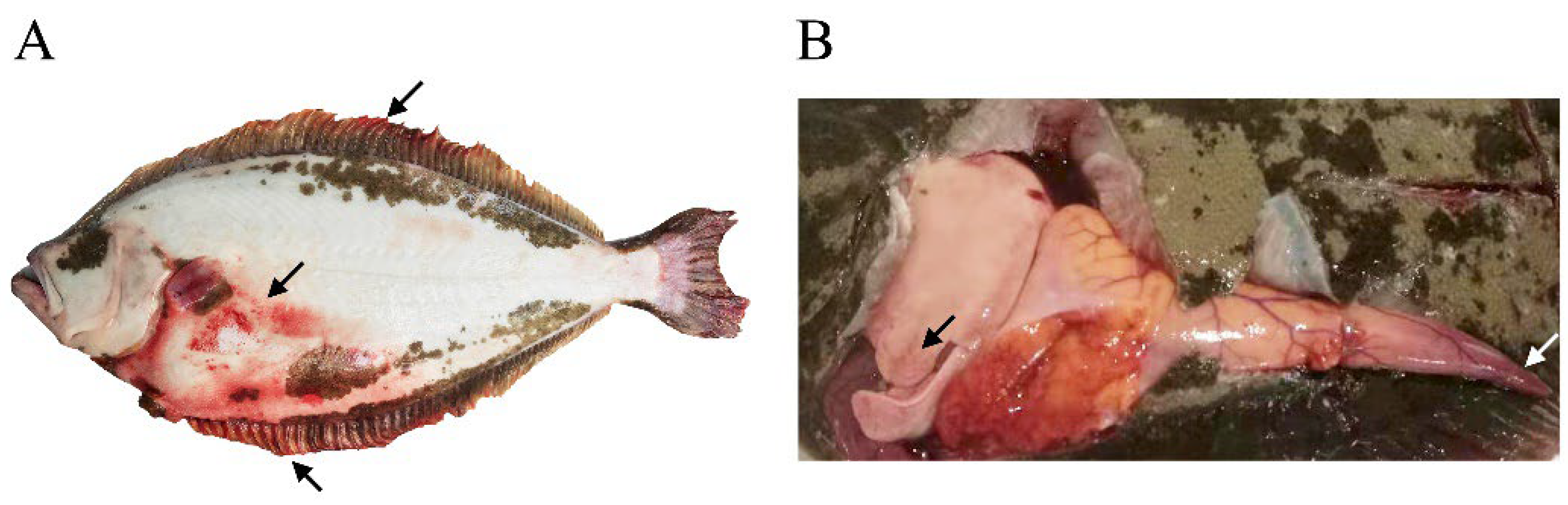
3.1. Pathology of Diseased Fish
3.2. Virus Isolation and Genome Sequence Analysis
3.3. RT-PCR Detection of Virus
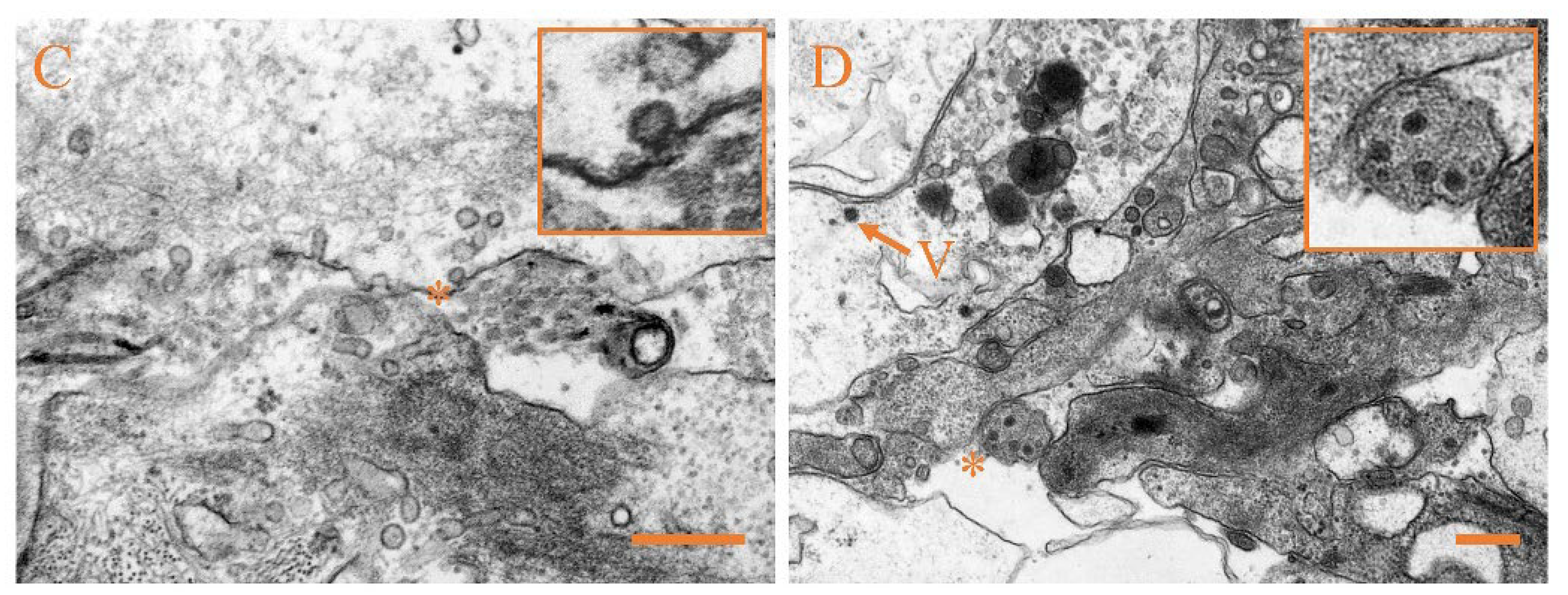
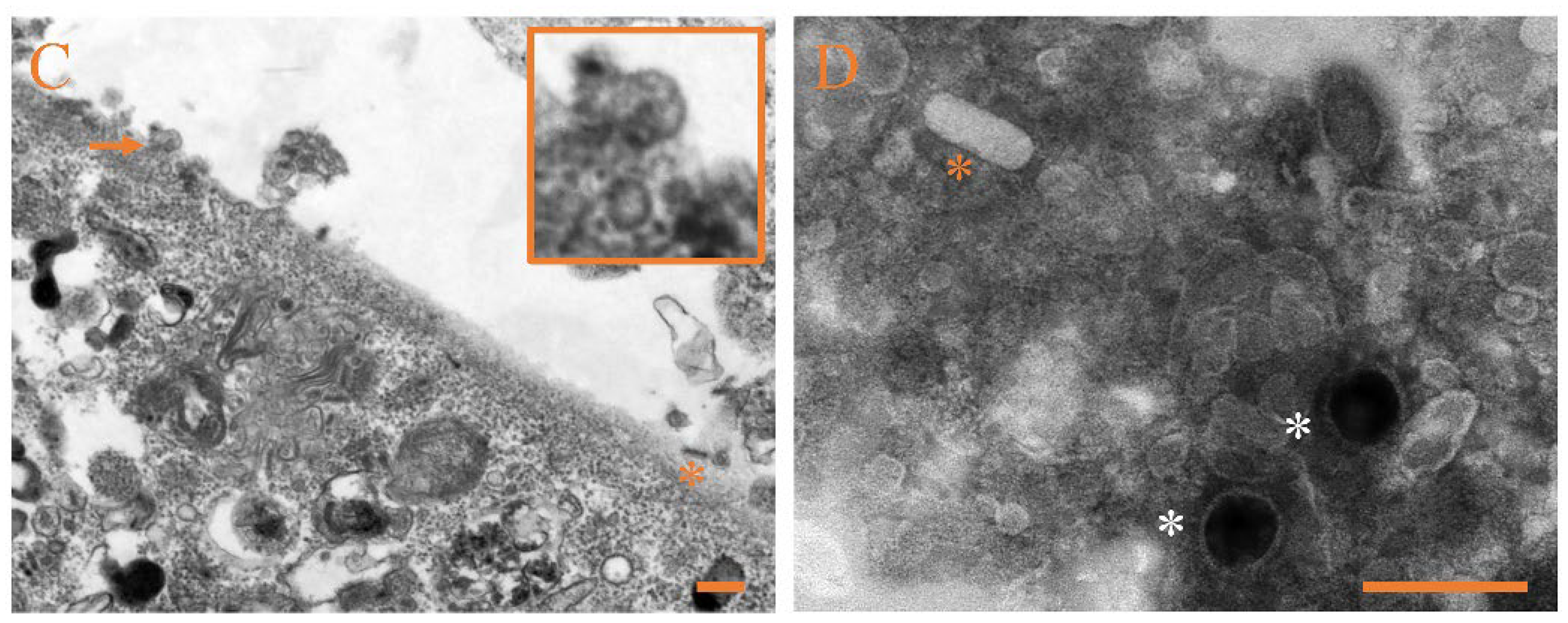
3.4. Ultrastructural Pathology and Characterization of Virion
3.5. RNA-Seq Results
3.6. Iso-Seq Results
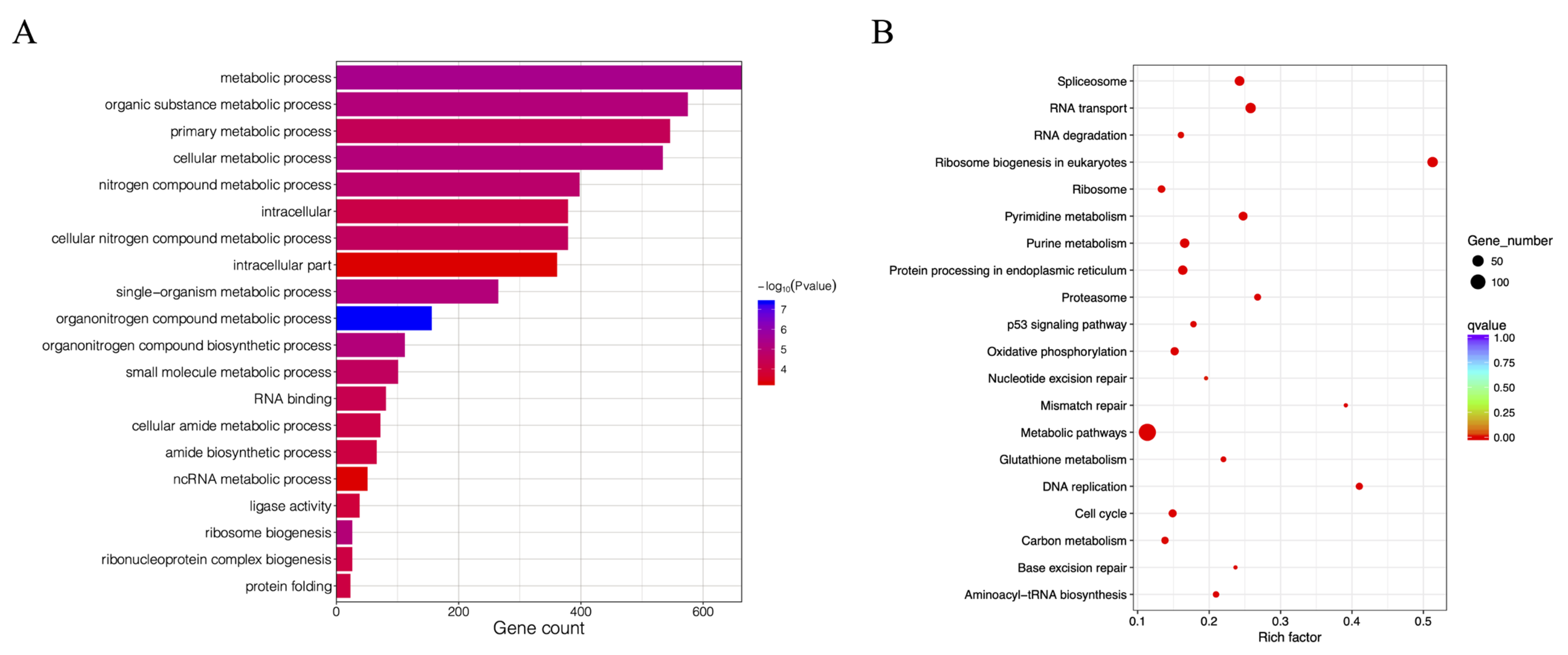
3.7. Analysis of DEGs
3.8. Detection of AS Events
3.9. Gene Alternative Splicing Is Involved in Signaling Pathways
3.10. Identification of TFs
3.11. LncRNA Predication
3.12. qRT-PCR Validation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, X.-Y.; Zhou, Y.; Chen, X.; Zheng, J.; Zeng, X.-D.; Ji, F. Isolation and genetic analysis of a nidovirus from crucian carp (Carassius auratus). Arch. Virol. 2019, 164, 1651–1654. [Google Scholar]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; et al. Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef]
- Cano, I.; Stone, D.; Savage, J.; Wood, G.; Mulhearn, B.; Gray, J.; Stinton, N.; Ross, S.; Bonar, M.; Taylor, N.G.H.; et al. Isolation of a Chinook Salmon Bafinivirus (CSBV) in Imported Goldfish Carassius auratus L. in the United Kingdom and Evaluation of Its Virulence in Resident Fish Species. Viruses 2020, 12, 578. [Google Scholar] [CrossRef]
- Granzow, H.; Weiland, F.; Fichtner, D.; Schütze, H.; Karger, A.; Mundt, E.; Dresenkamp, B.; Martin, P.; Mettenleiter, T.C. Identification and ultrastructural characterization of a novel virus from fish. J. Gen. Virol. 2001, 82, 2849–2859. [Google Scholar] [CrossRef] [Green Version]
- Iwanowicz, L.R.; Goodwin, A.E. A new bacilliform fathead minnow rhabdovirus that produces syncytia in tissue culture. Arch. Virol. 2002, 147, 899–915. [Google Scholar] [CrossRef]
- Lord, S.D.; Raymond, M.J.; Krell, P.J.; Kropinski, A.M.; Stevenson, R.M.W. Novel Chinook Salmon Bafnivirus Isolation from Ontario fish Health Monitoring. In Proceedings of the Seventh International Symposium on Aquatic Animal Health, Portland, OR, USA, 31 August–4 September 2014; p. 242. [Google Scholar]
- Zhang, X.; Shen, W.; Xu, C.; Wang, Y.; Xu, H.; Liu, X.; Wei, Y. Discovery of a novel Piscanivirus in yellow catfish (Pelteobagrus fulvidraco) in China. Infect. Genet. Evol. 2019, 74, 103924. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Chen, X.; Tian, J.-H.; Chen, L.-J.; Li, K.; Wang, W.; Eden, J.-S.; Shen, J.-J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Li, Z.; Liu, X.; Cheng, J.; He, Y.; Wang, X.; Wang, Z.; Qi, J.; Haiyang, Y.; Zhang, Q. Transcriptome profiling provides gene resources for understanding gill immune responses in Japanese flounder (Paralichthys olivaceus) challenged with Edwardsiella tarda. Fish Shellfish Immunol. 2017, 72, 593–603. [Google Scholar] [CrossRef]
- Wu, R.; Sheng, X.; Tang, X.; Xing, J.; Zhan, W. Transcriptome Analysis of Flounder (Paralichthys olivaceus) Gill in Response to Lymphocystis Disease Virus (LCDV) Infection: Novel Insights into Fish Defense Mechanisms. Int. J. Mol. Sci. 2018, 19, 160. [Google Scholar] [CrossRef] [Green Version]
- Nam, G.-H.; Mishra, A.; Gim, J.-A.; Lee, H.-E.; Jo, A.; Yoon, D.; Kim, A.; Kim, W.-J.; Ahn, K.; Kim, D.-H.; et al. Gene expression profiles alteration after infection of virus, bacteria, and parasite in the Olive flounder (Paralichthys olivaceus). Sci. Rep. 2018, 8, 18065. [Google Scholar] [CrossRef]
- Luo, D.; Zhou, Q.; Wu, Y.; Chai, X.; Liu, W.; Wang, Y.; Yang, Q.; Wang, Z.; Liu, Z. Full-length transcript sequencing and comparative transcriptomic analysis to evaluate the contribution of osmotic and ionic stress components towards salinity tolerance in the roots of cultivated alfalfa (Medicago sativa L.). BMC Plant Biol. 2019, 19, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.; Liu, H.; Zhang, J.; Yang, S.; Kong, G.; Chu, J.S.C.; Chen, N.; Wang, D. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genom. 2015, 16, 1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Nan, X.; Zhang, X.; Wang, G.; Ren, Y.; Wang, Y.; Fu, Y.; Hou, J. Molecular characterization and functional analysis of Japanese flounder (Paralichthys olivaceus) thbs2 in response to lymphocystis disease virus. Fish Shellfish Immunol. 2019, 93, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Wen, H.; Wang, H.; Lu, J.; Lu, L.; Jiang, Y. Identification of structure proteins of cyprinid herpesvirus 2. Aquaculture 2020, 523, 735184. [Google Scholar] [CrossRef]
- Wu, Q.; Lu, Y.; Lu, R.; Lau, N.; Lai, E.C.; Li, W.-X.; Ding, S.-W. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 1606–1611. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Liu, Y.; Hao, Y.; Liu, Y.; Wang, G.; He, Z.; Zhao, Y.; Xu, Z.; Liu, X.; Wang, Y.; Gong, C.; et al. atp6v0b gene regulates the immune response against Vibrio vulnificus in half-smooth tongue sole (Cynoglossus semilaevis). Aquac. Rep. 2021, 20, 100758. [Google Scholar] [CrossRef]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef]
- Shao, C.; Bao, B.; Xie, Z.; Chen, X.; Li, B.; Jia, X.; Yao, Q.; Ortí, G.; Li, W.; Li, X.; et al. The genome and transcriptome of Japanese flounder provide insights into flatfish asymmetry. Nat. Genet. 2017, 49, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Shi, M.; Wang, J.; Yang, N.; Wang, K.; Xi, J.; Wu, C.; Xi, T.; Zheng, J.; Zhang, J. Isoform Sequencing Provides Insight Into Freezing Response of Common Wheat (Triticum aestivum L.). Front. Genet. 2020, 11, 462. [Google Scholar] [CrossRef] [PubMed]
- Alamancos, G.P.; Pages, A.; Trincado, J.L.; Bellora, N.; Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 2015, 21, 1521–1531. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Miao, Y.-R.; Jia, L.-H.; Yu, Q.-Y.; Zhang, Q.; Guo, A.-Y. AnimalTFDB 3.0: A comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2019, 47, D33–D38. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Paul, T.P.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Nishizawa, T.; Furuhashi, M.; Nagai, T.; Nakai, T.; Muroga, K. Genomic classification of fish nodaviruses by molecular phylogenetic analysis of the coat protein gene. Appl. Environ. Microbiol. 1997, 63, 1633–1636. [Google Scholar] [CrossRef] [Green Version]
- Chi, S.C.; Shieh, J.R.; Lin, S.J. Genetic and antigenic analysis of betanodaviruses isolated from aquatic organisms in Taiwan. Dis. Aquat. Org. 2003, 55, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Amos, K.; Thomas, J.; Hopper, K. A Case History of Adaptive Management Strategies for Viral Hemorrhagic Septicemia Virus (VHSV) in Washington State. J. Aquat. Anim. Health 1998, 10, 152–159. [Google Scholar] [CrossRef]
- López-Vázquez, C.; Dopazo, C.P.; Barja, J.L.; Bandín, I. Experimental infection of turbot, Psetta maxima (L.), with strains of viral haemorrhagic septicaemia virus isolated from wild and farmed marine fish. J. Fish Dis. 2007, 30, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.Y.; Markkandan, K.; Kwon, M.G.; Seo, J.S.; Yoo, S.-I.; Hwang, S.D.; Son, M.-H.; Park, J. Transcriptome analysis of olive flounder (Paralichthys olivaceus) head kidney infected with moderate and high virulent strains of infectious viral hemorrhagic septicaemia virus (VHSV). Fish Shellfish Immunol. 2018, 76, 293–304. [Google Scholar] [CrossRef]
- Zhang, X.; Hou, J.; Wang, G.; Jiang, H.; Wang, Y.; Sun, Z.; Jiang, X.; Yu, Q.; Haijin Liu, H. Gonadal Transcriptome Analysis in Sterile Double Haploid Japanese Flounder. PLoS ONE 2015, 10, e0143204. [Google Scholar] [CrossRef] [Green Version]
- Boudreault, S.; Roy, P.; Lemay, G.; Bisaillon, M. Viral modulation of cellular RNA alternative splicing: A new key player in virus–host interactions? Wiley Interdiscip. Rev. RNA 2019, 10, e1543. [Google Scholar] [CrossRef] [Green Version]
- Troha, K.; Ayres, J.S. Metabolic adaptations to infections at the organismal level. Trends Immunol. 2020, 41, 113–125. [Google Scholar] [CrossRef]
- Yang, P.L. Metabolomics and lipidomics. In Viral Pathog; Elsevier: Amsterdam, The Netherlands, 2016; pp. 181–198. [Google Scholar]
- Malmstrøm, M.; Matschiner, M.; Tørresen, O.K.; Star, B.; Snipen, L.G.; Hansen, T.F.; Baalsrud, H.T.; Nederbragt, A.J.; Hanel, R.; Salzburger, W.; et al. Evolution of the immune system influences speciation rates in teleost fishes. Nat. Genet. 2016, 48, 1204–1212. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Chen, J.; Li, Y.; Wang, W.; Du, X.; Song, W.; Zhang, W.; Lin, L.; Yuan, Z. Hepatitis B Virus Polymerase Disrupts K63-Linked Ubiquitination of STING To Block Innate Cytosolic DNA-Sensing Pathways. J. Virol. 2015, 89, 2287–2300. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Tu, J.; Zhang, Y.; Wang, J.; Yang, L.; Wang, W.; Wu, Z.; Meng, Q.; Lin, L. Transcriptomic analysis of the head kidney of Topmouth culter (Culter alburnus) infected with Flavobacterium columnare with an emphasis on phagosome pathway. Fish Shellfish Immunol. 2016, 57, 413–418. [Google Scholar] [CrossRef]
- Li, S.; Chen, X.; Hao, G.; Geng, X.; Zhan, W.; Sun, J. Identification and characterization of a novel NOD-like receptor family CARD domain containing 3 gene in response to extracellular ATP stimulation and its role in regulating LPS-induced innate immune response in Japanese flounder (Paralichthys olivaceus) head kidney macrophages. Fish Shellfish Immunol. 2016, 50, 79–90. [Google Scholar] [PubMed]
- Li, Y.; Yuan, W.; Xu, Q.; Liu, H.; Dai, X. The regulation of immune responses against white spot syndrome virus or Vibrio alginolyticus in toll-like receptors silenced giant freshwater prawn (Macrobrachium rosenbergii). Fish Shellfish Immunol. 2020, 107, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Zheng, W.; Sun, Y.; Xu, T. microRNA-489 negatively modulates RIG-I signaling pathway via targeting TRAF6 in Miiuy croaker after poly(I:C) stimulation. Fish Shellfish Immunol. 2021, 113, 61–68. [Google Scholar] [CrossRef]
- Kuan, Y.-C.; Sheu, F.; Lee, G.-C.; Tsai, M.-W.; Hung, C.-L.; Nan, F.-H. Administration of recombinant Reishi immunomodulatory protein (rLZ-8) diet enhances innate immune responses and elicits protection against nervous necrosis virus in grouper Epinephelus coioides. Fish Shellfish Immunol. 2012, 32, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Lu, J.; Wang, Q.; Chen, S.; Xu, A.; Yuan, S. Autophagy participates in innate immune defense in lamprey. Fish Shellfish Immunol. 2018, 83, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-T.; Aoki, T.; Huang, Y.-T.; Hirono, I.; Chen, T.-C.; Huang, J.-Y.; Chang, G.-D.; Lo, C.-F.; Wang, H.-C. White Spot Syndrome Virus Induces Metabolic Changes Resembling the Warburg Effect in Shrimp Hemocytes in the Early Stage of Infection. J. Virol. 2011, 85, 12919–12928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.-J.; Yu, J.-W.; Wan, L.; Zhang, X.-Y.; Shi, Y.-G.; Chen, M.-Y. Endocannabinoid system activation contributes to glucose metabolism disorders of hepatocytes and promotes hepatitis C virus replication. Int. J. Infect. Dis. 2014, 23, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Su, M.-A.; Huang, Y.-T.; Chen, I.-T.; Lee, D.-Y.; Hsieh, Y.-C.; Li, C.-Y.; Ng, T.H.; Liang, S.-Y.; Lin, S.-Y.; Huang, S.-W.; et al. An Invertebrate Warburg Effect: A Shrimp Virus Achieves Successful Replication by Altering the Host Metabolome via the PI3K-Akt-mTOR Pathway. PLoS Pathog. 2014, 10, e1004196. [Google Scholar] [CrossRef] [Green Version]
- Delgado, T.; Carroll, P.A.; Punjabi, A.S.; Margineantu, D.; Hockenbery, D.M.; Lagunoff, M. Induction of the Warburg effect by Kaposi’s sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 10696–10701. [Google Scholar] [CrossRef] [Green Version]
- Delgado, T.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Global Metabolic Profiling of Infection by an Oncogenic Virus: KSHV Induces and Requires Lipogenesis for Survival of Latent Infection. PLoS Pathog. 2012, 8, e1002866. [Google Scholar] [CrossRef] [Green Version]
- Abrantes, J.L.; Alves, C.M.; Costa, J.; Almeida, F.C.L.; Sola-Penna, M.; Fontes, C.F.L.; Souz, T.M.L. Herpes simplex type 1 activates glycolysis through engagement of the enzyme 6-phosphofructo-1-kinase (PFK-1). Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1198–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allonso, D.; Andrade, I.S.; Conde, J.N.; Coelho, D.R.; Rocha, D.C.P.; da Silva, M.L.; Ventura, G.T.; Silva, E.M.; Mohana-Borges, R. Dengue Virus NS1 Protein Modulates Cellular Energy Metabolism by Increasing Glyceraldehyde-3-Phosphate Dehydrogenase Activity. J. Virol. 2015, 89, 11871–11883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakiri, D.; Minamitani, T.; Samanta, M. Epstein-Barr Virus Latent Membrane Protein 2A Contributes to Anoikis Resistance through ERK Activation. J. Virol. 2013, 87, 8227–8234. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Yu, L.; Fu, X.; Yan, X.; Lin, Q.; Liu, L.; Liang, H.; Li, N. iTRAQ-based proteomic profile analysis of ISKNV-infected CPB cells with emphasizing on glucose metabolism, apoptosis and autophagy pathways. Fish Shellfish Immunol. 2018, 79, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Green, R.E.; Brenner, S.E. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc. Natl. Acad. Sci. USA 2003, 100, 189–192. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Zhai, J.; Dang, S.; Zheng, S. Analysis of alternative splicing in chicken embryo fibroblasts in response to reticuloendotheliosis virus infection. Avian Pathol. 2018, 47, 585–594. [Google Scholar] [CrossRef]
- Hu, B.; Huo, Y.; Yang, L.; Chen, G.; Luo, M.; Yang, J.; Zhou, J. ZIKV infection effects changes in gene splicing, isoform composition and lncRNA expression in human neural progenitor cells. Virol. J. 2017, 14, 217. [Google Scholar] [CrossRef] [Green Version]
- Adair, R.; Liebisch, G.W.; Su, Y.; Colberg-Poley, A.M. Alteration of cellular RNA splicing and polyadenylation machineries during productive human cytomegalovirus infection. J. Gen. Virol. 2004, 85, 3541–3553. [Google Scholar] [CrossRef]
- Dowling, D.; Nasr-Esfahani, S.; Tan, C.H.; O’Brien, K.; Howard, J.L.; Jans, D.A.; Purcell, D.F.; Stoltzfus, C.M.; Sonza, S. HIV-1 infection induces changes in expression of cellular splicing factors that regulate alternative viral splicing and virus production in macrophages. Retrovirology 2008, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Boudreault, S.; Martenon-Brodeur, C.; Caron, M.; Garant, J.-M.; Tremblay, M.-P.; Armero, V.E.S.; Durand, M.; Lapointe, E.; Thibault, P.; Tremblay-Létourneau, M.; et al. Global Profiling of the Cellular Alternative RNA Splicing Landscape during Virus-Host Interactions. PLoS ONE 2016, 11, e0161914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davy, C.; Doorbar, J. G2/M cell cycle arrest in the life cycle of viruses. Virology 2007, 368, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Huang, Y.; Dai, M.; Zhao, X.; Du, Q.; Dong, F.; Wang, L.; Huo, R.; Zhang, W.; Xu, X.; et al. Transmissible gastroenteritis virus infection induces cell cycle arrest at S and G2/M phases via p53-dependent pathway. Virus Res. 2013, 178, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Wu, H.; Huang, J.; Xu, Y.; Yang, F.; Zhang, Q.; Xu, X. Porcine epidemic diarrhea virus through p53-dependent pathway causes cell cycle arrest in the G0/G1 phase. Virus Res. 2018, 253, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schlereth, K.; Beinoraviciute-Kellner, R.; Zeitlinger, M.K.; Bretz, A.C.; Sauer, M.; Charles, J.l.P.; Vogiatzi, F.; Leich, E.; Samans, B.; Eilers, M.; et al. DNA Binding Cooperativity of p53 Modulates the Decision between Cell-Cycle Arrest and Apoptosis. Mol. Cell 2010, 38, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | S | |
|---|---|---|
| Polymerase read bases (G) | 23.31 | 24.30 |
| Subreads base (G) | 22.43 | 23.62 |
| CCS | 450,095 | 435,572 |
| NFL | 59,771 | 44,964 |
| FLNC | 386,725 | 383,604 |
| FLNC/CCS | 85.92% | 88.07% |
| Polished consensus reads | 212,319 | 183,681 |
| Mean length of polished consensus reads | 2186 | 2922 |
| N50 of polished consensus reads | 2643 | 3429 |
| Pathways | DEGs Number |
|---|---|
| p53 signaling pathway | 13 |
| Phagosome | 17 |
| Cytosolic DNA-sensing pathway | 7 |
| NOD-like receptor signaling pathway | 7 |
| Salmonella infection | 9 |
| Toll-like receptor signaling pathway | 7 |
| Herpes simplex infection | 11 |
| RIG-I-like receptor signaling pathway | 4 |
| Lysosome | 8 |
| Apoptosis | 4 |
| Regulation of autophagy | 1 |
| Sample | Total Genes | SE | MX | RI | A5 | A3 | AF | AL | Total AS |
|---|---|---|---|---|---|---|---|---|---|
| R | 12,989 | 2298 (17.69%) | 234 (1.8%) | 3018 (23.24%) | 2219 (17.08%) | 2389 (18.39%) | 1049 (8.08%) | 245 (1.89%) | 11,452 |
| S | 10,743 | 2479 (23.08%) | 262 (2.44%) | 3358 (31.26%) | 2332 (21.71%) | 2590 (24.11%) | 1120 (10.43%) | 211 (1.96%) | 12,352 |
| Gene | AS in S | AS in R | Difference between S and R |
|---|---|---|---|
| tead3 | 27 | 52 | −25 |
| slco4a1 | 57 | 36 | 21 |
| sem4e | 13 | 34 | −21 |
| mrc1a | 28 | 9 | 19 |
| plcg2 | 28 | 9 | 19 |
| cttn | 2 | 20 | −18 |
| dgki | 12 | 27 | −15 |
| tktl1 | 7 | 22 | −15 |
| canx | 17 | 3 | 14 |
| sesn1 | 4 | 18 | −14 |
| atp8b2 | 27 | 14 | 13 |
| slc29a1 | 20 | 7 | 13 |
| mmp25 | 4 | 17 | −13 |
| hnrnpl | 17 | 6 | 11 |
| hspd1 | 13 | 2 | 11 |
| ssr1 | 9 | 20 | −11 |
| cpa1 | 1 | 12 | −11 |
| roaa | 15 | 5 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, C.; Zhang, Y.; Wang, G.; Liu, Y.; He, Z.; Ren, Y.; Cao, W.; Zhao, H.; Xu, Y.; Wang, Y.; et al. The Isolation and Full-Length Transcriptome Sequencing of a Novel Nidovirus and Response of Its Infection in Japanese Flounder (Paralichthys olivaceus). Viruses 2022, 14, 1216. https://doi.org/10.3390/v14061216
Gong C, Zhang Y, Wang G, Liu Y, He Z, Ren Y, Cao W, Zhao H, Xu Y, Wang Y, et al. The Isolation and Full-Length Transcriptome Sequencing of a Novel Nidovirus and Response of Its Infection in Japanese Flounder (Paralichthys olivaceus). Viruses. 2022; 14(6):1216. https://doi.org/10.3390/v14061216
Chicago/Turabian StyleGong, Chunguang, Yitong Zhang, Guixing Wang, Yufeng Liu, Zhongwei He, Yuqin Ren, Wei Cao, Haitao Zhao, Yuhao Xu, Yufen Wang, and et al. 2022. "The Isolation and Full-Length Transcriptome Sequencing of a Novel Nidovirus and Response of Its Infection in Japanese Flounder (Paralichthys olivaceus)" Viruses 14, no. 6: 1216. https://doi.org/10.3390/v14061216