
SARS-CoV-2 Causes Lung Inflammation through Metabolic Reprogramming and RAGE

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. SARS-CoV-2 Plasmids
2.2. Cell Culture and Transfection
2.3. Chemical Reagents
2.4. Western Blot Assay
2.5. RNA Extraction
2.6. qPCR Assay
2.7. Seahorse Mito Stress Test
2.8. Pyruvate Assay
2.9. Human Advanced Glycation End-Products (AGEs) ELISA
2.10. Cell Senescence Staining
2.11. Immunofluorescence Staining and Confocal Microscopy
2.12. Statistical Analysis
3. Results
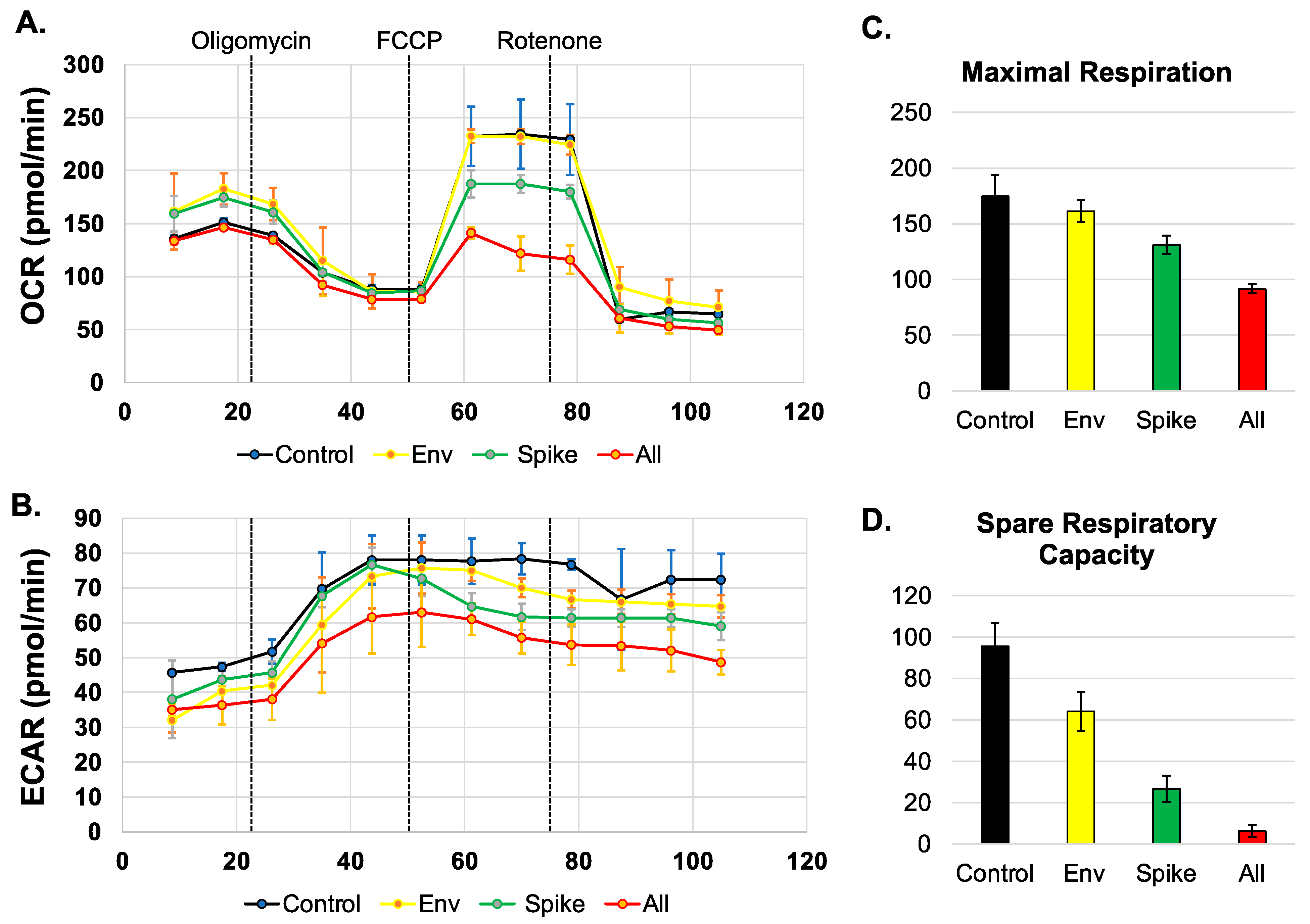
3.1. SARS-CoV-2 Proteins Disrupt the OCR
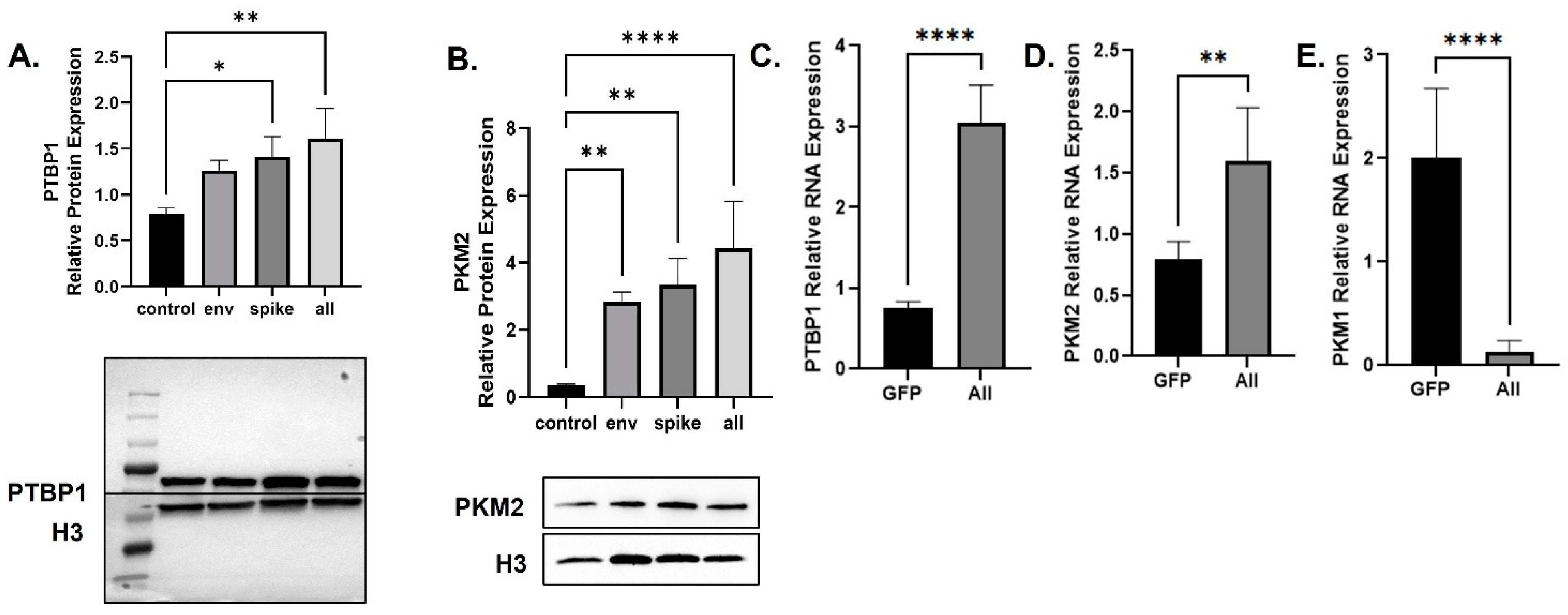
3.2. SARS-CoV-2 Increases PKM2 over PKM1
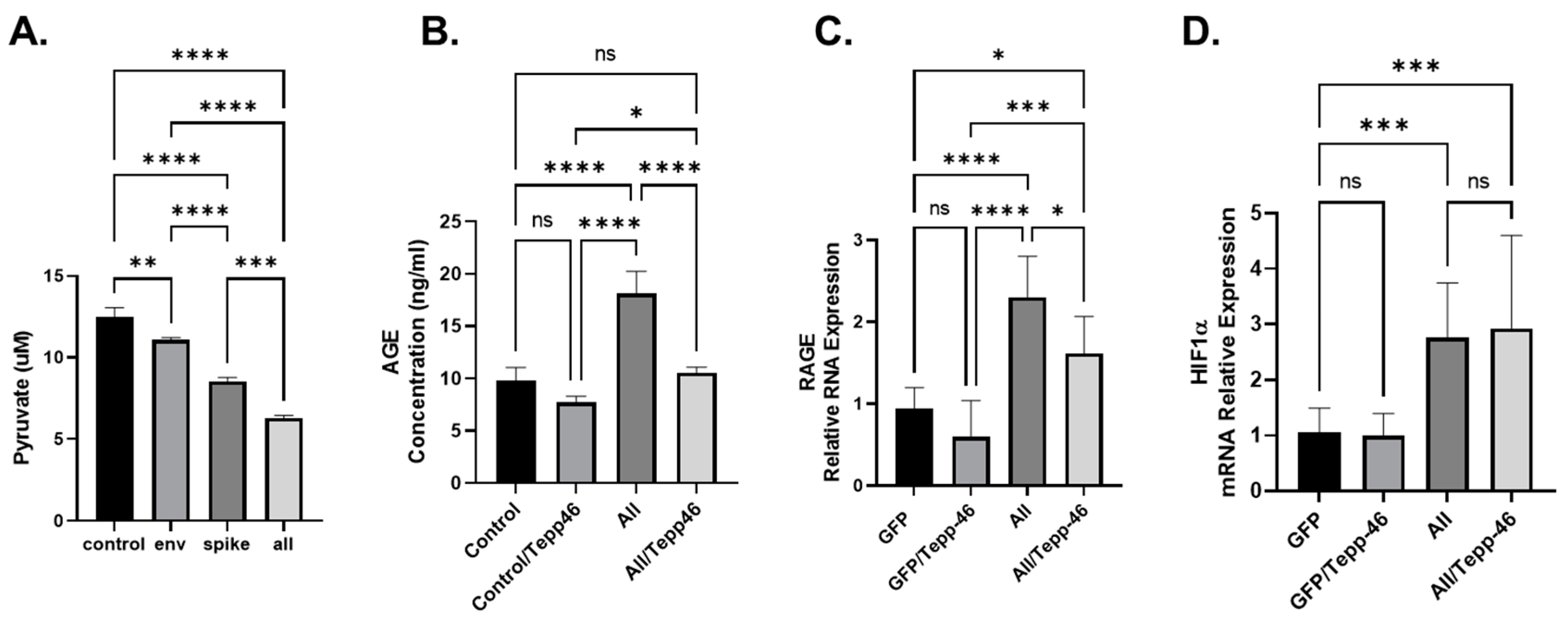
3.3. Tepp-46 Reverses the Effect of the SARS-CoV-2 Protein
3.4. SARS-CoV-2 Viral Proteins Result in Increased Advanced Glycation End-Products (AGEs)
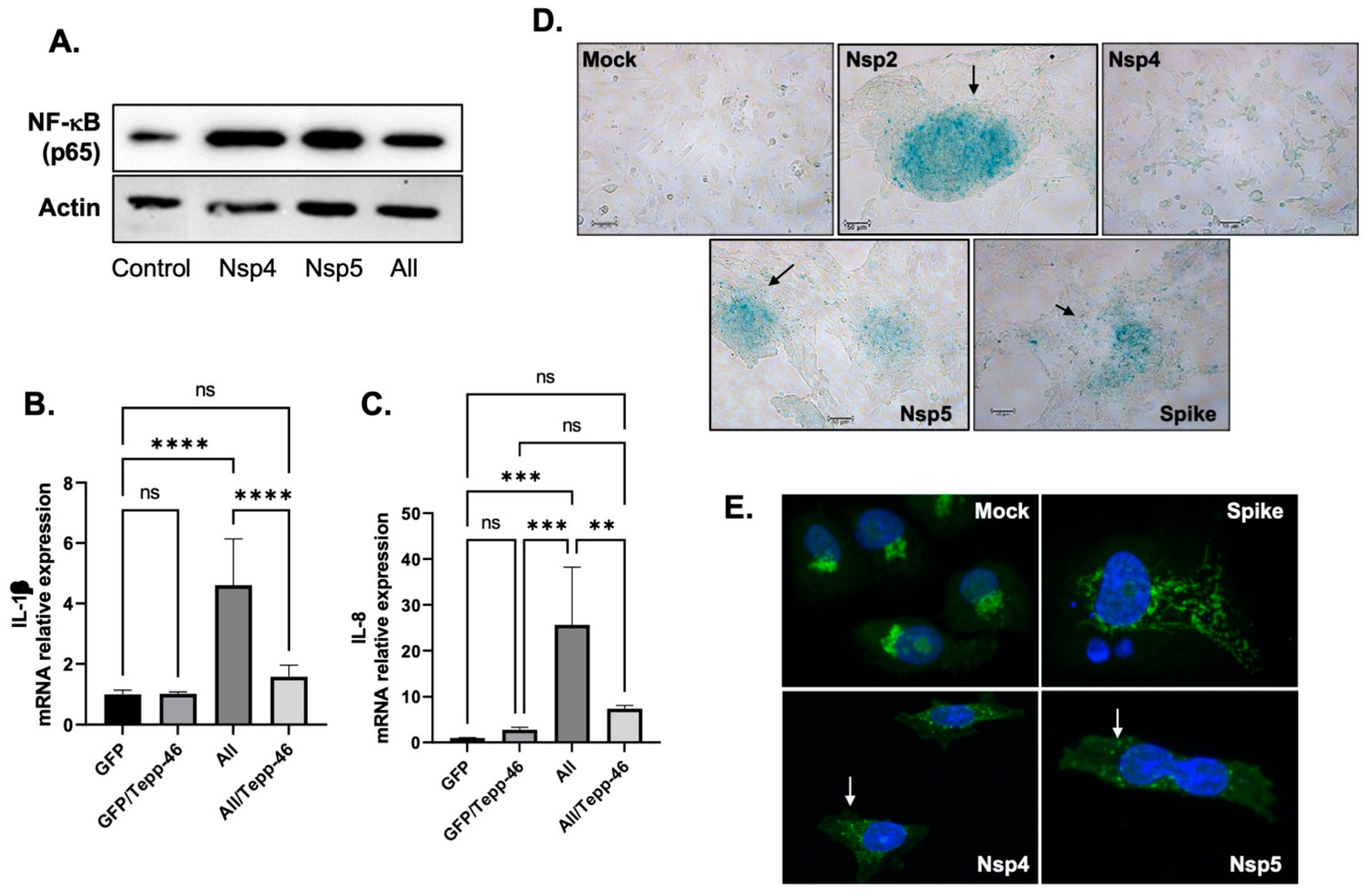
3.5. SARS-CoV-2 Proteins Induce Cytokines and Promote Cell Senescence
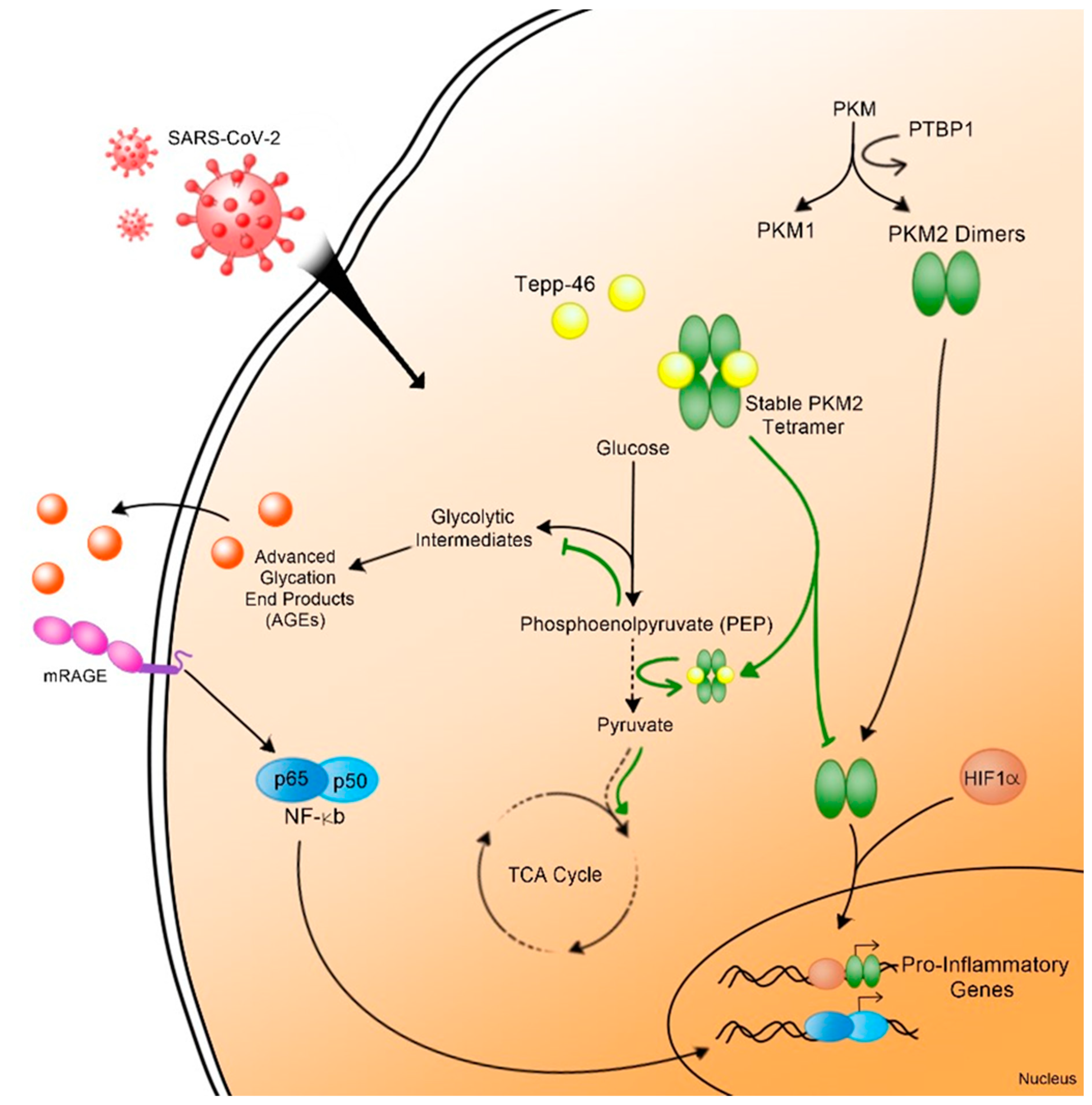
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Who Coronavirus (COVID-19) Dashboard; World Health Organization: Geneva, Switzerland, 2022; Available online: https://covid19.who.int/ (accessed on 30 March 2022).
- Mehandru, S.; Merad, M. Pathological sequelae of long-haul COVID. Nat. Immunol. 2022, 23, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Nie, M.; Pang, H.; Wang, B.; Hu, J.; Meng, X.; Li, K.; Ran, X.; Long, Q.; Deng, H.; et al. Integrated cytokine and metabolite analysis reveals immunometabolic reprogramming in COVID-19 patients with therapeutic implications. Nat. Commun. 2021, 12, 1618. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ma, F.; Yokota, T.; Garcia, G., Jr.; Palermo, A.; Wang, Y.; Farrell, C.; Wang, Y.-C.; Wu, R.; Zhou, Z.; et al. Metabolic reprogramming and epigenetic changes of vital organs in SARS-CoV-2–induced systemic toxicity. JCI Insight 2021, 6, e145027. [Google Scholar] [CrossRef]
- Han, R.; Liang, J.; Zhou, B. Glucose Metabolic Dysfunction in Neurodegenerative Diseases—New Mechanistic Insights and the Potential of Hypoxia as a Prospective Therapy Targeting Metabolic Reprogramming. Int. J. Mol. Sci. 2021, 22, 5887. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ning, J.; Cao, W.; Wang, S.; Du, T.; Jiang, J.; Feng, X.; Zhang, B. Research Progress of TXNIP as a Tumor Suppressor Gene Participating in the Metabolic Reprogramming and Oxidative Stress of Cancer Cells in Various Cancers. Front. Oncol. 2020, 10, 568574. [Google Scholar] [CrossRef]
- Ganesh, G.V.; Mohanram, R.K. Metabolic reprogramming and immune regulation in viral diseases. Rev. Med. Virol. 2021, 32, e2268. [Google Scholar] [CrossRef]
- Cassina, P.; Miquel, E.; Martínez-Palma, L.; Cassina, A. Glial Metabolic Reprogramming in Amyotrophic Lateral Sclerosis. Neuroimmunomodulation 2021, 28, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhou, W.; Dou, F.; Wang, C.; Yu, Z. TRPV1 sustains microglial metabolic reprogramming in Alzheimer’s disease. EMBO Rep. 2021, 22, e52013. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2021, 1–43. [Google Scholar] [CrossRef]
- Polcicova, K.; Badurova, L.; Tomaskova, J. Metabolic reprogramming as a feast for virus replication. Acta Virol. 2020, 64, 201–215. [Google Scholar] [CrossRef]
- Kubicka, A.; Matczak, K.; Łabieniec-Watała, M. More Than Meets the Eye Regarding Cancer Metabolism. Int. J. Mol. Sci. 2021, 22, 9507. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojkova, D.; Costa, R.; Bechtel, M.; Ciesek, R.; Michaelis, M.; Cinatl, J., Jr. Targeting pentose phosphate pathway for SARS-CoV-2 therapy. Metabolites 2021, 11, 699. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, A.K.; Chernukhin, I.; Bechtold, U. Profiling of advanced glycation end products uncovers abiotic stress-specific target proteins in Arabidopsis. J. Exp. Bot. 2018, 70, 653–670. [Google Scholar] [CrossRef] [PubMed]
- Puckett, D.L.; Alquraishi, M.; Chowanadisai, W.; Bettaieb, A. The Role of PKM2 in Metabolic Reprogramming: Insights into the Regulatory Roles of Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 1171. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Bamezai, R.N. Human pyruvate kinase M2: A multifunctional protein. Protein Sci. 2010, 19, 2031–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boschert, V.; Teusch, J.; Müller-Richter, U.D.A.; Brands, R.C.; Hartmann, S. PKM2 Modulation in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 775. [Google Scholar] [CrossRef]
- Satyanarayana, G.; Turaga, R.C.; Sharma, M.; Wang, S.; Mishra, F.; Peng, G.; Deng, X.; Yang, J.; Liu, Z.-R. Pyruvate kinase M2 regulates fibrosis development and progression by controlling glycine auxotrophy in myofibroblasts. Theranostics 2021, 11, 9331–9341. [Google Scholar] [CrossRef]
- Yumnamcha, T.; Guerra, M.; Singh, L.P.; Ibrahim, A.S. Metabolic Dysregulation and Neurovascular Dysfunction in Diabetic Retinopathy. Antioxidants 2020, 9, 1244. [Google Scholar] [CrossRef]
- Monu; Agnihotri, P.; Biswas, S. AGE/Non-AGE Glycation: An Important Event in Rheumatoid Arthritis Pathophysiology. Inflammation 2021, 45, 477–496. [Google Scholar] [CrossRef]
- Kerkeni, M.; Gharbi, J. RAGE receptor: May be a potential inflammatory mediator for SARS-CoV-2 infection? Med. Hypotheses 2020, 144, 109950. [Google Scholar] [CrossRef] [PubMed]
- Riehl, A.; Németh, J.; Angel, P.; Hess, J. The receptor RAGE: Bridging inflammation and cancer. Cell Commun. Signal. 2009, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. The multiple faces of RAGE—Opportunities for therapeutic intervention in aging and chronic disease. Expert Opin. Ther. Targets 2015, 20, 431–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Feng, Y.; Ouyang, Y.; Liang, Y.; Zhao, K.; Wang, Y.; Luo, Q.; Xiao, J.; Feng, N.; Zhou, M. Inhibition of advanced glycation endproducts formation by lotus seedpod oligomeric procyanidins through RAGE-MAPK signaling and NF-κB activation in high-AGEs-diet mice. Food Chem. Toxicol. 2021, 156, 112481. [Google Scholar] [CrossRef]
- Gopal, P.; Gosker, H.R.; Theije, C.C.; Eurlings, I.M.; Sell, D.R.; Monnier, V.M.; Reynaert, N.L. Effect of chronic hypoxia on RAGE and its soluble forms in lungs and plasma of mice. Biochim. Biophys. Acta 2015, 1852, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senatus, L.; MacLean, M.; Arivazhagan, L.; Egaña-Gorroño, L.; López-Díez, R.; Manigrasso, M.B.; Ruiz, H.H.; Vasquez, C.; Wilson, R.; Shekhtman, A.; et al. Inflammation Meets Metabolism: Roles for the Receptor for Advanced Glycation End Products Axis in Cardiovascular Disease. Immunometabolism 2021, 3, e210024. [Google Scholar] [CrossRef]
- Xu, L.; Wang, Y.-R.; Li, P.-C.; Feng, B. Advanced glycation end products increase lipids accumulation in macrophages through upregulation of receptor of advanced glycation end products: Increasing uptake, esterification and decreasing efflux of cholesterol. Lipids Health Dis. 2016, 15, 161. [Google Scholar] [CrossRef] [Green Version]
- Pratte, K.A.; Curtis, J.L.; Kechris, K.; Couper, D.; Cho, M.H.; Silverman, E.K.; DeMeo, D.L.; Sciurba, F.C.; Zhang, Y.; Ortega, V.E.; et al. Soluble receptor for advanced glycation end products (sRAGE) as a biomarker of COPD. Respir. Res. 2021, 22, 127. [Google Scholar] [CrossRef]
- Hughes, M.J.; McGettrick, H.M.; Sapey, E. Shared mechanisms of multimorbidity in COPD, atherosclerosis and type-2 diabetes: The neutrophil as a potential inflammatory target. Eur. Respir. Rev. 2020, 29, 190102. [Google Scholar] [CrossRef] [Green Version]
- Kehribar, D.Y.; Cihangiroglu, M.; Sehmen, E.; Avci, B.; Capraz, A.; Bilgin, A.Y.; Gunaydin, C.; Ozgen, M. The receptor for advanced glycation end product (RAGE) pathway in COVID-19. Biomarkers 2021, 26, 114–118. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Yu, F.; Yu, W.; Ye, P.; Liu, P.; Gu, Y.; Chen, S.; Zhang, H. Pyruvate kinase M2 activation protects against the proliferation and migration of pulmonary artery smooth muscle cells. Cell Tissue Res. 2020, 382, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Jennings, P.; Toma, C.; Gardner, I.; van de Water, B.; Forsby, A.; Leist, M. Development of a neurotoxicity assay that is tuned to detect mitochondrial toxicants. Arch. Toxicol. 2019, 93, 1585–1608. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Santerre, M.; Tempera, I.; Martin, K.; Mukerjee, R.; Sawaya, B.E. HIV-1 Vpr disrupts mitochondria axonal transport and accelerates neuronal aging. Neuropharmacology 2017, 117, 364–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, M.D.; Johnsen, I.B.; Stiberg, K.A.; Sherstova, T.; Wakita, T.; Richard, G.M.; Kandasamy, R.K.; Meurs, E.F.; Anthonsen, M.W. Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc. Natl. Acad. Sci. USA 2017, 114, E3462–E3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Ma, Y.; Liu, Y.; Wan, Q. Measurement of mitochondrial respiration in adherent cells by Seahorse XF96 Cell Mito Stress Test. STAR Protoc. 2020, 2, 100245. [Google Scholar] [CrossRef]
- Sansbury, B.E.; Jones, S.P.; Riggs, D.W.; Darley-Usmar, V.M.; Hill, B.G. Bioenergetic function in cardiovascular cells: The importance of the reserve capacity and its biological regulation. Chem. Biol. Interact. 2011, 191, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Yetkin-Arik, B.; Vogels, I.M.C.; Nowak-Sliwinska, P.; Weiss, A.; Houtkooper, R.H.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci. Rep. 2019, 9, 12608. [Google Scholar] [CrossRef] [Green Version]
- Thaker, S.K.; Ch’Ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 59. [Google Scholar] [CrossRef]
- Choksi, A.; Parulekar, A.; Pant, R.; Shah, V.K.; Nimma, R.; Firmal, P.; Singh, S.; Kundu, G.C.; Shukla, S.; Chattopadhyay, S. Tumor suppressor SMAR1 regulates PKM alternative splicing by HDAC6-mediated deacetylation of PTBP1. Cancer Metab. 2021, 9, 16. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, J.; Manley, J. Turning on a Fuel Switch of Cancer: hnRNP Proteins Regulate Alternative Splicing of Pyruvate Kinase mRNA. Cancer Res. 2010, 70, 8977–8980. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Uchiyama, K.; Akao, Y. PTBP1-targeting microRNAs regulate cancer-specific energy metabolism through the modulation of PKM1/M2 splicing. Cancer Sci. 2020, 112, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.N.; Arjona, S.P.; Santerre, M.; De Lucia, C.; Koch, W.J.; Sawaya, B.E. Metabolic Reprogramming in HIV-Associated Neurocognitive Disorders. Front. Cell. Neurosci. 2022, 16, 812887. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.L.; Khadka, V.; Tang, M.; Avelar, A.; Schunke, K.; Menor, M.; Shohet, R.V. HIF1 mediates a switch in pyruvate kinase isoforms after myocardial infarction. Physiol. Genom. 2018, 50, 479–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, C.N.S.; Arjona, S.P.; Santerre, M.; Sawaya, B.E. Hallmarks of Metabolic Reprogramming and Their Role in Viral Pathogenesis. Viruses 2022, 14, 602. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.A.; Wu, C.H.; Lu, C.C.; Hsia, S.M.; Yen, G.C. Glycative stress from advanced glycation end products (AGEs) and dicarbonyls: An emerging biological factor in cancer onset and progression. Mol. Nutr. Food Res. 2016, 60, 1850–1864. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Rai, V.; Frolov, S.; Singer, D.; Chabierski, S.; Xie, J.; Reverdatto, S.; Burz, D.S.; Schmidt, A.M.; Hoffman, R.; et al. Advanced glycation end product (AGE) recognition by the receptor for AGEs (RAGE). Structure 2011, 19, 722–732. [Google Scholar] [CrossRef] [Green Version]
- Holms, R.D. The COVID-19 Cell Signalling Problem: Spike, RAGE, PKC, p38, NFκB & IL-6 Hyper-Expression and the Human Ezrin Peptide, VIP, PKA-CREB Solution. Immuno 2022, 2, 260–282. [Google Scholar] [CrossRef]
- Cho, J.-H.; Saini, D.K.; Karunarathne, W.A.; Kalyanaraman, V.; Gautam, N. Alteration of Golgi structure in senescent cells and its regulation by a G protein γ subunit. Cell. Signal. 2011, 23, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Casals, M.; Brito-Zerón, P.; Mariette, X. Systemic and organ-specific immune-related manifestations of COVID-19. Nat. Rev. Rheumatol. 2021, 17, 315–332. [Google Scholar] [CrossRef]
- Tserel, L.; Jõgi, P.; Naaber, P.; Maslovskaja, J.; Häling, A.; Salumets, A.; Zusinaite, E.; Soeorg, H.; Lättekivi, F.; Ingerainen, D.; et al. Long-Term Elevated Inflammatory Protein Levels in Asymptomatic SARS-CoV-2 Infected Individuals. Front. Immunol. 2021, 12, 709759. [Google Scholar] [CrossRef] [PubMed]
- Michelen, M.; Manoharan, L.; Elkheir, N.; Cheng, V.; Dagens, A.; Hastie, C.; O’Hara, M.; Suett, J.; Dahmash, D.; Bugaeva, P.; et al. Characterising long COVID: A living systematic review. BMJ Glob. Health 2021, 6, e005427. [Google Scholar] [CrossRef] [PubMed]
- Montani, D.; Savale, L.; Noel, N.; Meyrignac, O.; Colle, R.; Gasnier, M.; Corruble, E.; Beurnier, A.; Jutant, E.-M.; Pham, T.; et al. Post-acute COVID-19 syndrome. Eur. Respir. Rev. 2022, 31, 210185. [Google Scholar] [CrossRef] [PubMed]
- Behzadi, F.; Ulloa, N.A.; Danckers, M. Multisystem inflammatory syndrome in adults: A case report and review of the literature. J. Med. Case Rep. 2022, 16, 102. [Google Scholar] [CrossRef] [PubMed]
- Borella, R.; De Biasi, S.; Paolini, A.; Boraldi, F.; Tartaro, D.L.; Mattioli, M.; Fidanza, L.; Neroni, A.; Caro-Maldonado, A.; Meschiari, M.; et al. Metabolic reprograming shapes neutrophil functions in severe COVID-19. Eur. J. Immunol. 2021, 52, 484–502. [Google Scholar] [CrossRef]
- Jia, H.; Liu, C.; Li, D.; Huang, Q.; Liu, D.; Zhang, Y.; Ye, C.; Zhou, D.; Wang, Y.; Tan, Y.; et al. Metabolomic analyses reveal new stage-specific features of COVID-19. Eur. Respir. J. 2021, 59, 2100284. [Google Scholar] [CrossRef]
- Mussap, M.; Fanos, V. Could metabolomics drive the fate of COVID-19 pandemic? A narrative review on lights and shadows. Clin. Chem. Lab. Med. 2021, 59, 1891–1905. [Google Scholar] [CrossRef]
- Ripoli, M.; D’Aprile, A.; Quarato, G.; Sarasin-Filipowicz, M.; Gouttenoire, J.; Scrima, R.; Cela, O.; Boffoli, D.; Heim, M.H.; Moradpour, D.; et al. Hepatitis C Virus-Linked Mitochondrial Dysfunction Promotes Hypoxia-Inducible Factor 1α-Mediated Glycolytic Adaptation. J. Virol. 2010, 84, 647–660. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Clippinger, A.J.; Alwine, J.C. Viral effects on metabolism: Changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol. 2011, 19, 360–367. [Google Scholar] [CrossRef] [Green Version]
- McFadden, K.; Hafez, A.Y.; Kishton, R.; Messinger, J.E.; Nikitin, P.A.; Rathmell, J.C.; Luftig, M.A. Metabolic stress is a barrier to Epstein–Barr virus-mediated B-cell immortalization. Proc. Natl. Acad. Sci. USA 2016, 113, E782–E790. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.; Mateus, M.; Thinnes, C.C.; McCullagh, J.; Schofield, C.J.; Taylor, G.P.; Bangham, C.R. Glucose Metabolism and Oxygen Availability Govern Reactivation of the Latent Human Retrovirus HTLV-1. Cell Chem. Biol. 2017, 24, 1377–1387.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passos, F.; Heimfarth, L.; Monteiro, B.S.; Corrêa, C.B.; de Moura, T.R.; Araújo, A.; Martins-Filho, P.R.; Quintans-Júnior, L.J.; Quintans, J. Oxidative stress and inflammatory markers in patients with COVID-19: Potential role of RAGE, HMGB1, GFAP and COX-2 in disease severity. Int. Immunopharmacol. 2022, 104, 108502. [Google Scholar] [CrossRef] [PubMed]
- Leisman, D.E.; Mehta, A.; Thompson, B.T.; Charland, N.C.; Gonye, A.; Gushterova, I.; Kays, K.R.; Khanna, H.K.; LaSalle, T.J.; Lavin-Parsons, K.M.; et al. Alveolar, Endothelial, and Organ Injury Marker Dynamics in Severe COVID-19. Am. J. Respir. Crit. Care Med. 2022, 205, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Tleyjeh, I.M.; Saddik, B.; AlSwaidan, N.; AlAnazi, A.; Ramakrishnan, R.K.; Alhazmi, D.; Aloufi, A.; AlSumait, F.; Berbari, E.; Halwani, R. Prevalence and predictors of Post-Acute COVID-19 Syndrome (PACS) after hospital discharge: A cohort study with 4 months median follow-up. PLoS ONE 2021, 16, e0260568. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allen, C.N.S.; Santerre, M.; Arjona, S.P.; Ghaleb, L.J.; Herzi, M.; Llewellyn, M.D.; Shcherbik, N.; Sawaya, B.E. SARS-CoV-2 Causes Lung Inflammation through Metabolic Reprogramming and RAGE. Viruses 2022, 14, 983. https://doi.org/10.3390/v14050983
Allen CNS, Santerre M, Arjona SP, Ghaleb LJ, Herzi M, Llewellyn MD, Shcherbik N, Sawaya BE. SARS-CoV-2 Causes Lung Inflammation through Metabolic Reprogramming and RAGE. Viruses. 2022; 14(5):983. https://doi.org/10.3390/v14050983
Chicago/Turabian StyleAllen, Charles N. S., Maryline Santerre, Sterling P. Arjona, Lea J. Ghaleb, Muna Herzi, Megan D. Llewellyn, Natalia Shcherbik, and Bassel E. Sawaya. 2022. "SARS-CoV-2 Causes Lung Inflammation through Metabolic Reprogramming and RAGE" Viruses 14, no. 5: 983. https://doi.org/10.3390/v14050983