
Analysis of Coxsackievirus B5 Infections in the Central Nervous System in Brazil: Insights into Molecular Epidemiology and Genetic Diversity

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Background and Virus Isolation
2.2. Molecular Detection and Sequencing
2.3. Phylogenetic Analysis
2.4. In Silico Analysis
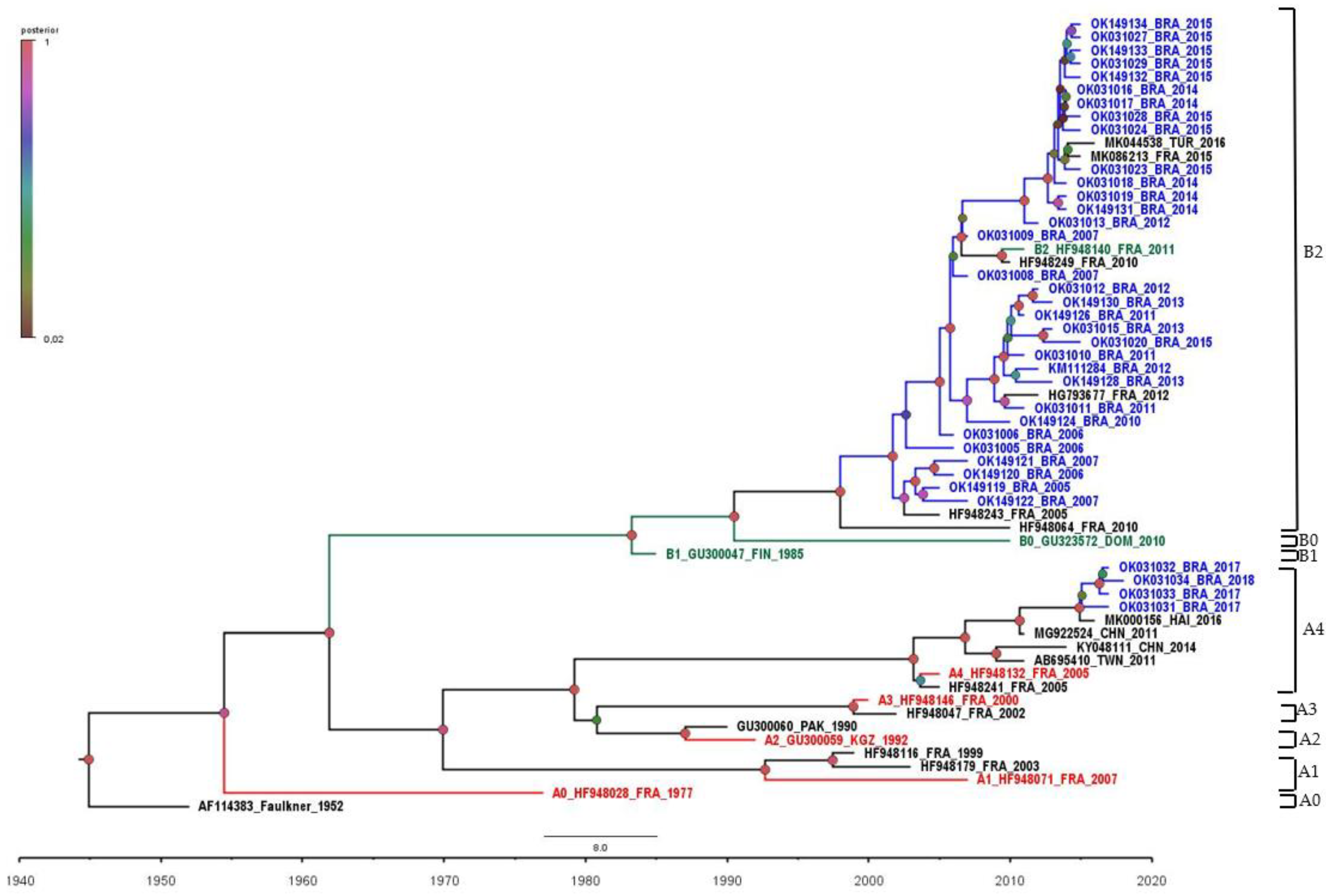
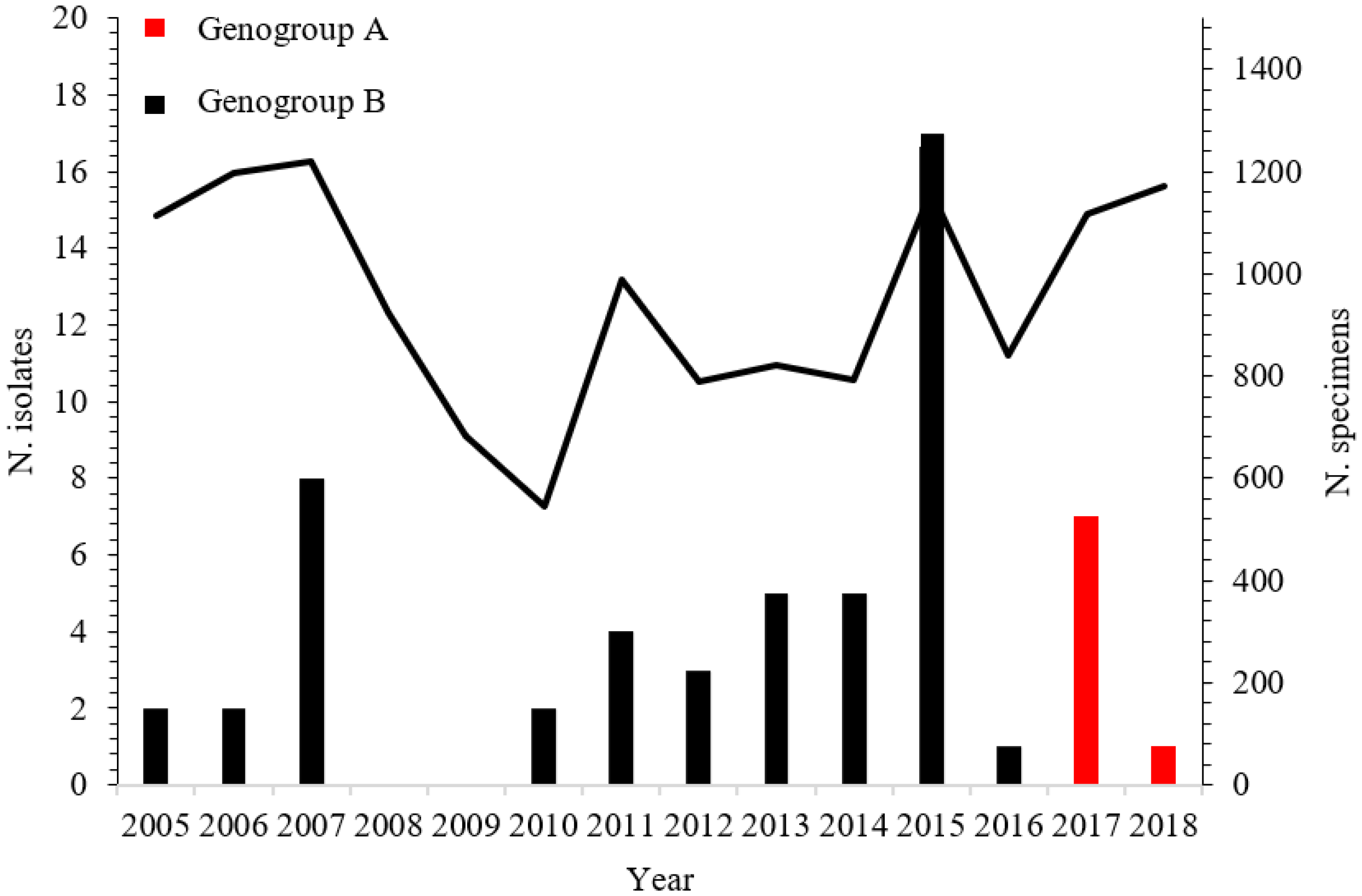
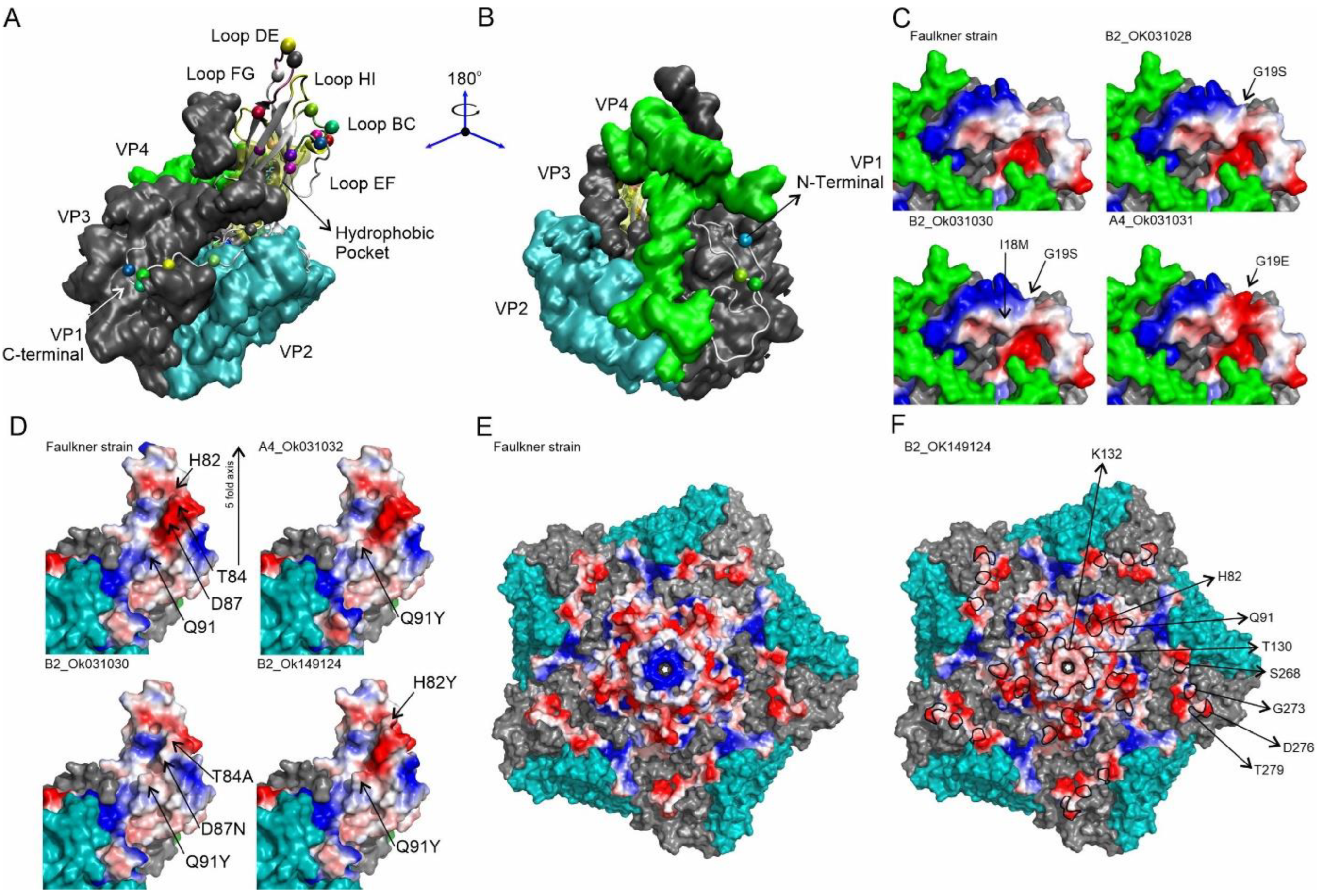
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A6S | Replacement of alanine by serine at position 6 of the VP1 protein |
| A90G | Replacement of alanine by glycine at position 90 of the VP1 protein |
| A90T | Replacement of alanine by threonine at position 90 of the VP1 protein |
| D87N | Replacement of aspartic acid by asparagine at position 87 of the VP1 protein |
| D276E | Replacement of aspartic acid by glutamic acid at position 276 of the VP1 protein |
| G4E | Replacement of glycine by glutamic acid at position 4 of the VP1 protein |
| G19E | Replacement of glycine by glutamic acid at position 19 of the VP1 protein |
| G19S | Replacement of glycine by serine at position 19 of the VP1 protein |
| G273S | Replacement of glycine by serine at position 273 of the VP1 protein |
| H82Y | Replacement of histidine by tyrosine at position 82 of the VP1 protein |
| I7V | Replacement of isoleucine by valine at position 7 of the VP1 protein |
| I18M | Replacement of isoleucine by methionine at position 18 of the VP1 protein |
| K132Q | Replacement of lysine by glutamine at position 132 of the VP1 protein |
| M180I | Replacement of methionine by isoleucine at position 180 of the VP1 protein |
| N95S | Replacement of asparagine by serine at position 95 of the VP1 protein |
| N262S | Replacement of asparagine by serine at position 262 of the VP1 protein |
| Q91Y | Replacement of glutamine by tyrosine at position 91 of the VP1 protein |
| Q258E | Replacement of glutamine by glutamic acid at position 258 of the VP1 protein |
| R9Q | Replacement of arginine by glutamine at position 9 of the VP1 protein |
| R200K | Replacement of arginine by lysine at position 200 of the VP1 protein |
| S125T | Replacement of serine by threonine at position 125 of the VP1 protein |
| S136A | Replacement of serine by alanine at position 136 of the VP1 protein |
| S268T | Replacement of serine by threonine at position 268 of the VP1 protein |
| T84A | Replacement of threonine by alanine at position 84 of the VP1 protein |
| T130A | Replacement of threonine by alanine at position 130 of the VP1 protein |
| T130N | Replacement of threonine by asparagine at position 130 of the VP1 protein |
| T275I | Replacement of threonine by isoleucine at position 275 of the VP1 protein |
| T279A | Replacement of threonine by alanine at position 279 of the VP1 protein |
| V156I | Replacement of valine by isoleucine at position 156 of the VP1 protein |
| V235I | Replacement of valine by isoleucine at position 235 of the VP1 protein |
| V235A | Replacement of valine by alanine at position 235 of the VP1 protein |
| V248A | Replacement of valine by alanine at position 248 of the VP1 protein |
References
- Koyuncu, O.O.; Hogue, I.B.; Enquist, L.W. Virus infections in the nervous system. Cell Host Microbe 2013, 13, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ICTV. Available online: https://talk.ictvonline.org/ (accessed on 14 February 2022).
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet. Evol. 2013, 14, 282–293. [Google Scholar] [CrossRef]
- Sousa, I.P., Jr.; Maximo, A.C.B.; Figueiredo, M.A.A.; Maia, Z.; da Silva, E.E. Echovirus 30 detection in an outbreak of acute myalgia and rhabdomyolysis, Brazil 2016–2017. Clin. Microbiol. Infect. 2019, 25, 252.e5–252.e8. [Google Scholar] [CrossRef] [PubMed]
- Pallansch, M.A.; Roos, R.P. Enteroviruses: Polioviruses, Coxsackieviruses, Echoviruses, and Newer Enteroviruses. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Willians & Wilkins: Philadelphia, PA, USA, 2001; pp. 723–776. [Google Scholar]
- Liu, N.; Jia, L.; Yin, J.; Wu, Z.; Wang, Z.; Li, P.; Hao, R.; Wang, L.; Wang, Y.; Qiu, S.; et al. An outbreak of aseptic meningitis caused by a distinct lineage of coxsackievirus B5 in China. Int. J. Infect. Dis. 2014, 23, 101–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalho, E.; Sousa, I., Jr.; Burlandy, F.; Costa, E.; Dias, A.; Serrano, R.; Oliveira, M.; Lopes, R.; Debur, M.; Burger, M.; et al. Identification and Phylogenetic Characterization of Human Enteroviruses Isolated from Cases of Aseptic Meningitis in Brazil, 2013–2017. Viruses 2019, 11, 690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, I.P., Jr.; Oliveira, M.L.A.; Burlandy, F.M.; Machado, R.S.; Oliveira, S.S.; Tavares, F.N.; Gomes-Neto, F.; da Costa, E.V.; da Silva, E.E. Molecular characterization and epidemiological aspects of non-polio enteroviruses isolated from acute flaccid paralysis in Brazil: A historical series (2005–2017). Emerg. Microbes Infect. 2020, 9, 2536–2546. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, G.P.; da Costa, E.V.; Tavares, F.N.; da Costa, L.J.; da Silva, E.E. Genetic diversity of Echovirus 30 involved in aseptic meningitis cases in Brazil (1998–2008). J. Med. Virol. 2011, 83, 2164–2171. [Google Scholar] [CrossRef] [PubMed]
- Rezig, D.; Fares, W.; Seghier, M.; Yahia, A.B.; Touzi, H.; Triki, H. Update on molecular characterization of coxsackievirus B5 strains. J. Med. Virol. 2011, 83, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Henquell, C.; Mirand, A.; Richter, J.; Schuffenecker, I.; Böttiger, B.; Diedrich, S.; Terletskaia-Ladwig, E.; Christodoulou, C.; Peigue-Lafeuille, H.; Bailly, J.L. Phylogenetic patterns of human coxsackievirus B5 arise from population dynamics between two genogroups and reveal evolutionary factors of molecular adaptation and transmission. J. Virol. 2013, 87, 12249–12259. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Huang, X.; Kang, K.; Li, X.; Tang, X.; Ren, Y.; Wang, Y.; Zhao, G.; Xu, B. Recombination in human coxsackievirus B5 strains that caused an outbreak of viral encephalitis in Henan, China. Arch. Virol. 2013, 158, 2169–2173. [Google Scholar] [CrossRef] [PubMed]
- Shen, H. Recombination analysis of coxsackievirus B5 genogroup C. Arch. Virol. 2018, 163, 539–544. [Google Scholar] [CrossRef]
- Mirand, A.; Archimbaud, C.; Henquell, C.; Michel, Y.; Chambon, M.; Peigue-Lafeuille, H.; Bailly, J. Prospective identification of HEV-B enteroviruses during the 2005 outbreak. J. Med. Virol. 2006, 78, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA 7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchard, M.; Lemey, P.; Baele, G.; Ayre, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Nicholls, G.K.; Rodrigo, A.G.; Solomon, W. Estimating mutation parameters population history and genealogy simultaneously from temporally spaced sequence data. Genetics 2002, 161, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Minin, V.N.; Bloomquist, E.W.; Suchard, M.A. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol. Biol. Evol. 2008, 25, 1459–1471. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Hall, T.A. BIOEDIT: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamyatnin, A.A. Protein volume in solution. Prog. Biophys. Mol. Biol. 1972, 24, 107–123. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–28. [Google Scholar] [CrossRef]
- Yuan, J.; Shen, L.; Wu, J.; Zou, X.; Gu, J.; Chen, J.; Mao, L. Enterovirus A71 Proteins: Structure and Function. Front. Microbiol. 2018, 9, 286. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Liu, Y.; Ma, H.C.; Paul, A.V.; Wimmer, E. Picornavirus morphogenesis. Microbiol. Mol. Biol. Rev. 2014, 78, 418–437. [Google Scholar] [CrossRef] [Green Version]
- Minor, P.D.; Schild, G.C.; Bootman, J.; Evans, D.M.; Ferguson, M.; Reeve, P.; Spitz, M.; Stanway, G.; Cann, A.J.; Hauptmann, R.; et al. Location and primary structure of a major antigenic site for poliovirus neutralization. Nature 1983, 301, 674–679. [Google Scholar] [CrossRef]
- Minor, P.D.; Ferguson, M.; Evans, D.M.; Almond, J.W.; Icenogle, J.P. Antigenic structure of polioviruses of serotypes 1, 2 and 3. J. Gen. Virol. 1986, 67, 1283–1291. [Google Scholar] [CrossRef]
- Fernandez-Garcia, M.D.; Kebe, O.; Fall, A.D.; Ndiaye, K. Identification and molecular characterization of non-polio enteroviruses from children with acute flaccid paralysis in West Africa, 2013–2014. Sci. Rep. 2017, 7, 3808. [Google Scholar] [CrossRef]
- Kapoor, A.; Victoria, J.; Simmonds, P.; Slikas, E.; Chieochansin, T.; Naeem, A.; Shaukat, S.; Sharif, S.; Alam, M.M.; Angez, M.; et al. A highly prevalent and genetically diversified Picornaviridae genus in South Asian children. Proc. Natl. Acad. Sci. USA 2008, 105, 20482–20487. [Google Scholar] [CrossRef] [Green Version]
- Sousa, I.P., Jr.; Dos Santos, F.B.; de Paula, V.S.; Vieira, T.C.R.G.; Dias, H.G.; Barros, C.A.; da Silva, E.E. Viral and Prion Infections Associated with Central Nervous System Syndromes in Brazil. Viruses 2021, 13, 1370. [Google Scholar] [CrossRef]
- Huang, H.W.; Chu, P.H.; Pan, C.H.; Wang, C.F.; Lin, C.C.; Lu, P.L.; Chen, Y.S.; Shi, Y.Y.; Su, H.J.; Chou, L.C.; et al. Evolutionary histories of coxsackievirus B5 and swine vesicular disease virus reconstructed by phylodynamic and sequence variation analyses. Sci. Rep. 2018, 8, 8821. [Google Scholar] [CrossRef] [Green Version]
- Angez, M.; Shaukat, S.; Zahra, R.; Khurshid, A.; Sharif, S.; Alam, M.M.; Zaidi, S.S. Molecular epidemiology of enterovirus B77 isolated from non polio acute flaccid paralytic patients in Pakistan during 2013. Infect. Genet. Evol. 2015, 29, 189–195. [Google Scholar] [CrossRef]
- Oberste, M.S. Comparative genomics of the coxsackie B viruses and related enteroviruses. Curr. Top. Microbiol. Immunol. 2008, 323, 33–47. [Google Scholar]
- Cifuente, J.O.; Moratorio, G. Evolutionary and Structural Overview of Human Picornavirus Capsid Antibody Evasion. Front. Cell Infect. Microbiol. 2019, 9, 283. [Google Scholar] [CrossRef]
- Battistone, A.; Buttinelli, G.; Fiore, S.; Amato, C.; Bonomo, P.; Patti, A.M.; Vulcano, A.; Barbi, M.; Binda, S.; Pellegrinelli, L.; et al. Sporadic isolation of sabin-like polioviruses and high-level detection of non-polio enteroviruses during sewage surveillance in seven Italian cities, after several years of inactivated poliovirus vaccination. Appl. Environ. Microbiol. 2014, 80, 4491–4501. [Google Scholar] [CrossRef] [Green Version]
- Norder, H.; Bjerregaard, L.; Magnius, L.; Lina, B.; Aymard, M.; Chomel, J.J. Sequencing of ’untypable’ enteroviruses reveals two new types, EV-77 and EV-78, within human enterovirus type B and substitutions in the BC loop of the VP1 protein for known types. J. Gen. Virol. 2003, 84, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Cordey, S.; Petty, T.J.; Schibler, M.; Martinez, Y.; Gerlach, D.; van Belle, S.; Turin, L.; Zdobnov, E.; Kaiser, L.; Tapparel, C. Identification of site-specific adaptations conferring increased neural cell tropism during human enterovirus 71 infection. PLoS Pathog. 2012, 8, e1002826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashev, A.N.; Vakulenko, Y.A. Molecular evolution of types in non-polio enteroviruses. J. Gen. Virol. 2017, 98, 2968–2981. [Google Scholar] [CrossRef] [PubMed]
- Ang, P.Y.; Chong, C.W.H.; Alonso, S. Viral determinants that drive Enterovirus-A71 fitness and virulence. Emerg. Microbes Infect. 2021, 10, 713–724. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VP1 Amino Acid Residue | Prototype Strain Residue | Genogroup A | Genogroup B | PROVEAN Prediction | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | Location | A0 | A1 | A2 | A3 | A4 | B0 | B1 | B2 | ||
| 4 | N-terminus | G | G4E | Neutral | |||||||
| 6 | N-terminus | A | A6S | Neutral | |||||||
| 7 | N-terminus | I | I7V | I7V | Neutral | ||||||
| 9 | N-terminus | R | R9Q | Neutral | |||||||
| 18 | N-terminus | I | I18M | Neutral | |||||||
| 19 | N-terminus | G | G19E | G19S | Neutral | ||||||
| 82 | BC-loop | H | H82Y | Neutral | |||||||
| 84 | BC-loop | T | T84A | Neutral | |||||||
| 87 | BC-loop | D | D87N | Neutral | |||||||
| 90 | BC-loop | A | A90G | A90T | Neutral | ||||||
| 91 | βC-strand | Q | Q91Y | Q91Y | Neutral | ||||||
| 95 | CD-loop | N | N95S | Neutral | |||||||
| 125 | βD-strand | S | S125T | S125T | Neutral | ||||||
| 130 | DE-loop | T | T130A/T130N | Deleterious | |||||||
| 132 | DE-loop | K | K132Q | K132Q | Neutral | ||||||
| 136 | DE-loop | S | S136A | Neutral | |||||||
| 156 | EF-loop | V | V156I | Neutral | |||||||
| 158 | EF-loop | S | S158C | Neutral | |||||||
| 169 | βF-strand | V | V169I | Neutral | |||||||
| 180 | βG-strand | M | M180I | Neutral | |||||||
| 200 | βH-αF | R | R200K | Neutral | |||||||
| 235 | βJ-strand | V | V235I | V235A | Neutral | ||||||
| 248 | C-terminus | V | V248A | Neutral | |||||||
| 258 | C-terminus | Q | Q258E | Neutral | |||||||
| 262 | C-terminus | N | N262S | Neutral | |||||||
| 268 | C-terminus | S | S268T | S268T | Neutral | ||||||
| 273 | C-terminus | G | G273S | G273S | Neutral | ||||||
| 275 | C-terminus | T | T275I | Neutral | |||||||
| 276 | C-terminus | D | D276E | Neutral | |||||||
| 279 | C-terminus | T | T279A | Neutral | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, R.S.; Gomes-Neto, F.; Aguiar-Oliveira, M.L.; Burlandy, F.M.; Tavares, F.N.; da Silva, E.E.; Sousa, I.P., Jr. Analysis of Coxsackievirus B5 Infections in the Central Nervous System in Brazil: Insights into Molecular Epidemiology and Genetic Diversity. Viruses 2022, 14, 899. https://doi.org/10.3390/v14050899
Machado RS, Gomes-Neto F, Aguiar-Oliveira ML, Burlandy FM, Tavares FN, da Silva EE, Sousa IP Jr. Analysis of Coxsackievirus B5 Infections in the Central Nervous System in Brazil: Insights into Molecular Epidemiology and Genetic Diversity. Viruses. 2022; 14(5):899. https://doi.org/10.3390/v14050899
Chicago/Turabian StyleMachado, Raiana S., Francisco Gomes-Neto, Maria L. Aguiar-Oliveira, Fernanda M. Burlandy, Fernando N. Tavares, Edson E. da Silva, and Ivanildo P. Sousa, Jr. 2022. "Analysis of Coxsackievirus B5 Infections in the Central Nervous System in Brazil: Insights into Molecular Epidemiology and Genetic Diversity" Viruses 14, no. 5: 899. https://doi.org/10.3390/v14050899