
Human Norovirus Induces Aquaporin 1 Production by Activating NF-κB Signaling Pathway
 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses, Cell Lines, Antibodies and Inhibitors
2.2. Plasmid Construction
2.3. Luciferase Reporter Assay
2.4. RNA Isolation and Quantitative PCR
2.5. Immunofluorescence Assay (IFA)
2.6. Western Blot (WB)
2.7. HIE (Human Intestinal Enteroids) Construction
2.7.1. Human Crypts Isolation
2.7.2. Three-Dimensional (3D) Culture of HIEs
2.7.3. HIE Passage and Differentiation
2.8. Human IEB (Intestinal Epithelial Barrier) Construction and Application
2.8.1. IEB Construction
2.8.2. Trans-Epithelial Electrical Resistance (TEER)
2.8.3. Paracellular Permeability
2.9. Negative Staining for Transmission Electron Microscopy
2.10. Statistical Analysis
3. Results
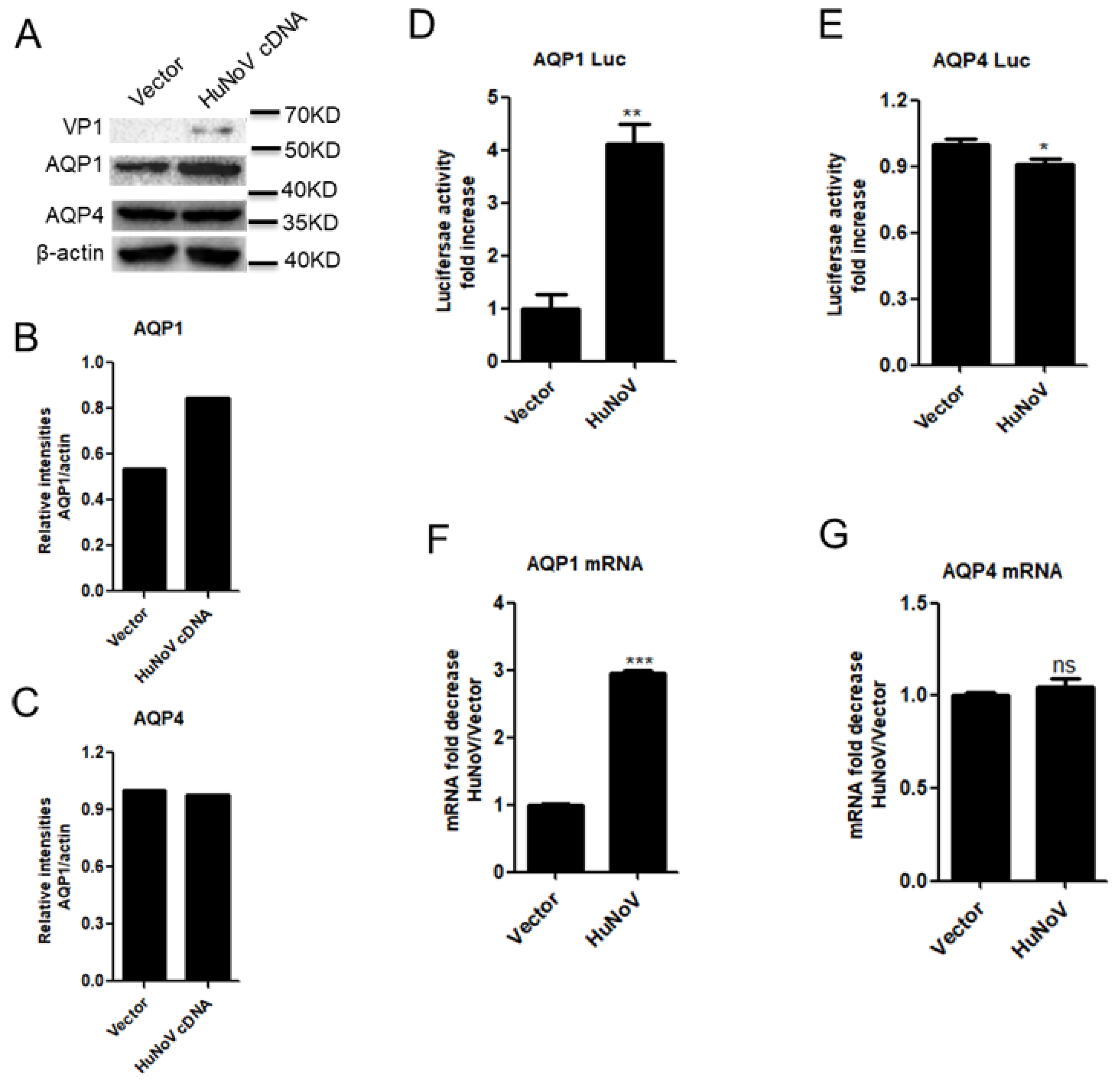
3.1. HuNoV Transfection Promotes AQP1 Expression
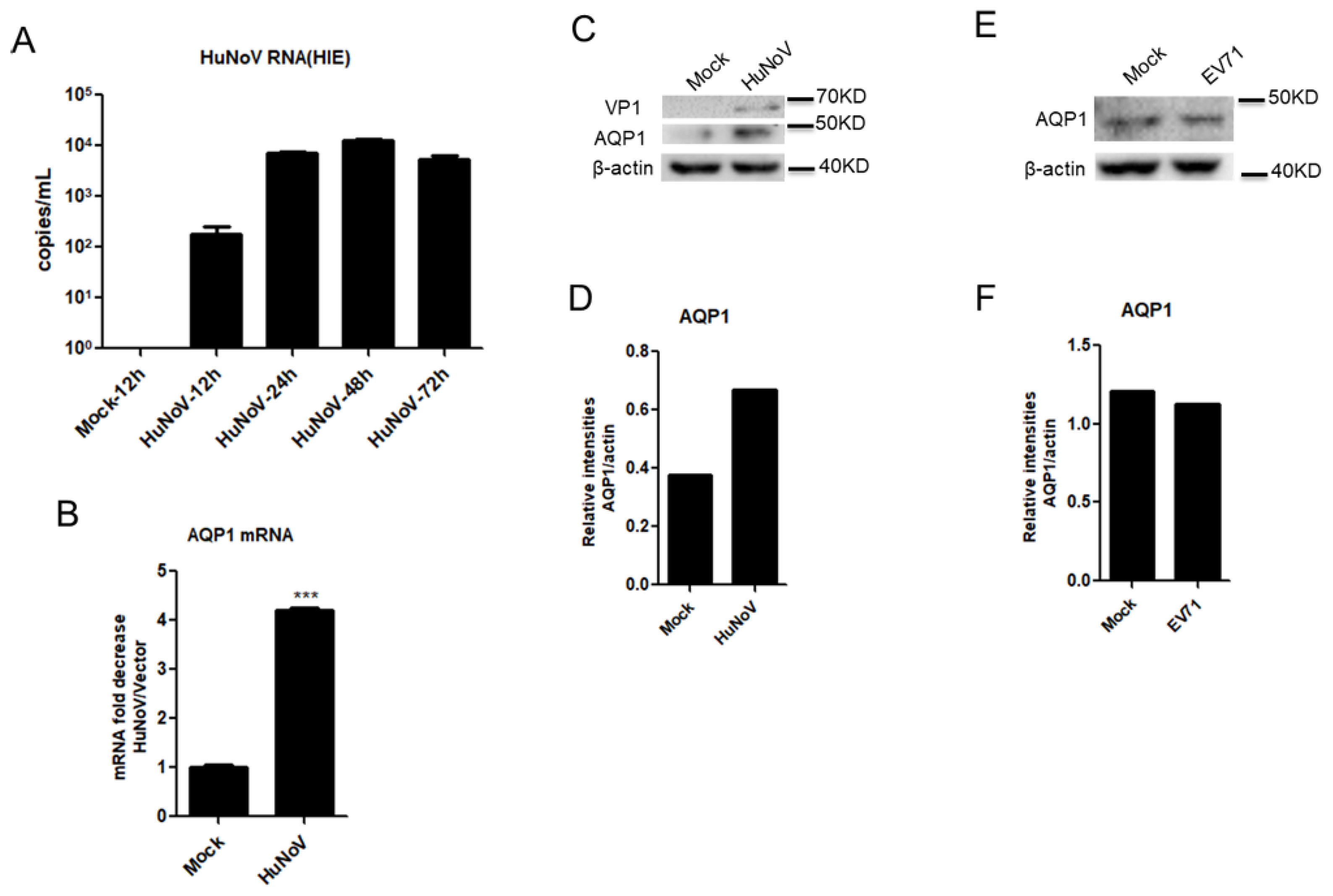
3.2. HuNoV Infection Promotes AQP1 Expression
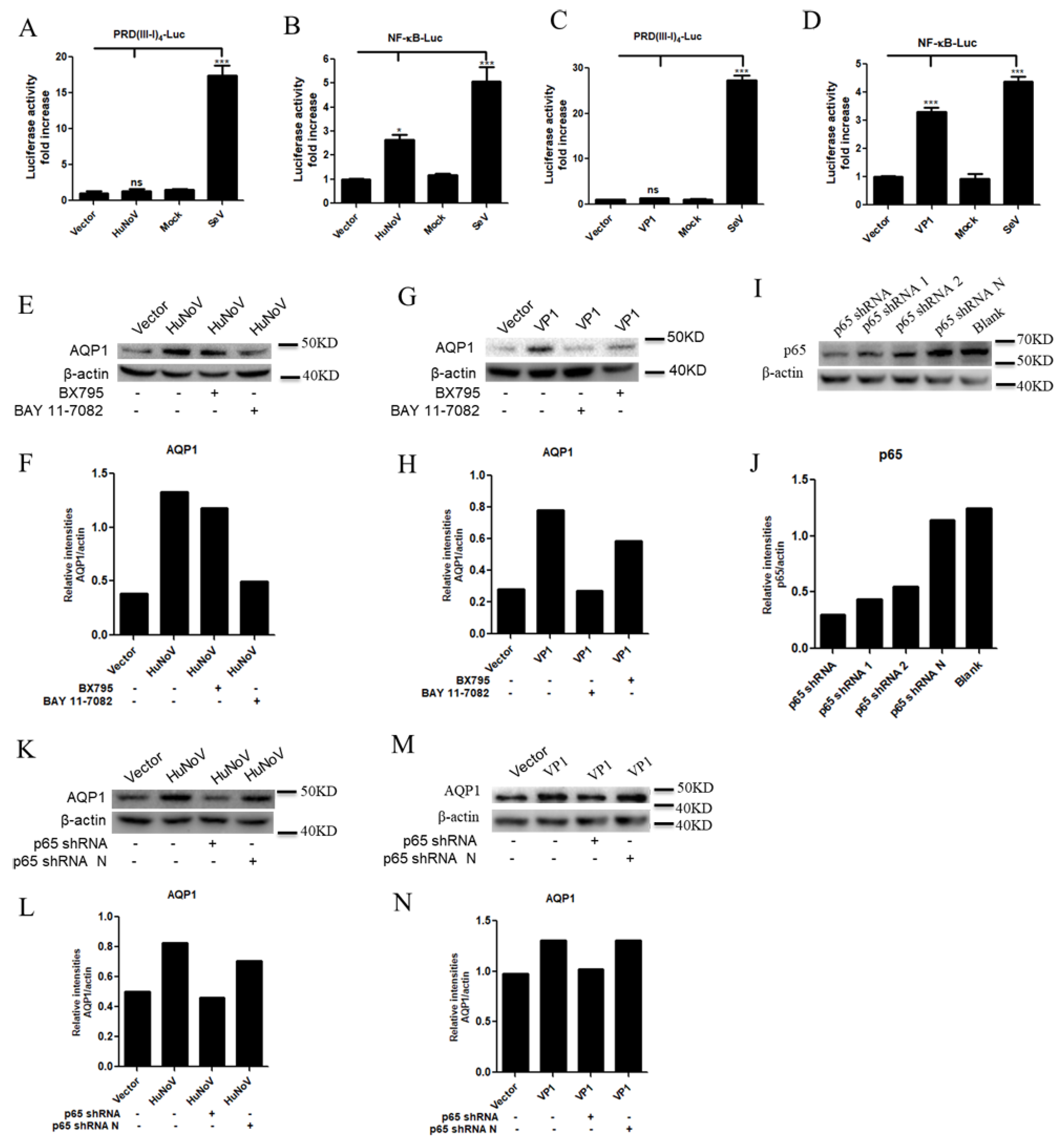
3.3. HuNoV VP1 Promotes the Expression of AQP1
3.4. HuNoV Promotes AQP1 Expression through NF-κB Signaling Pathway
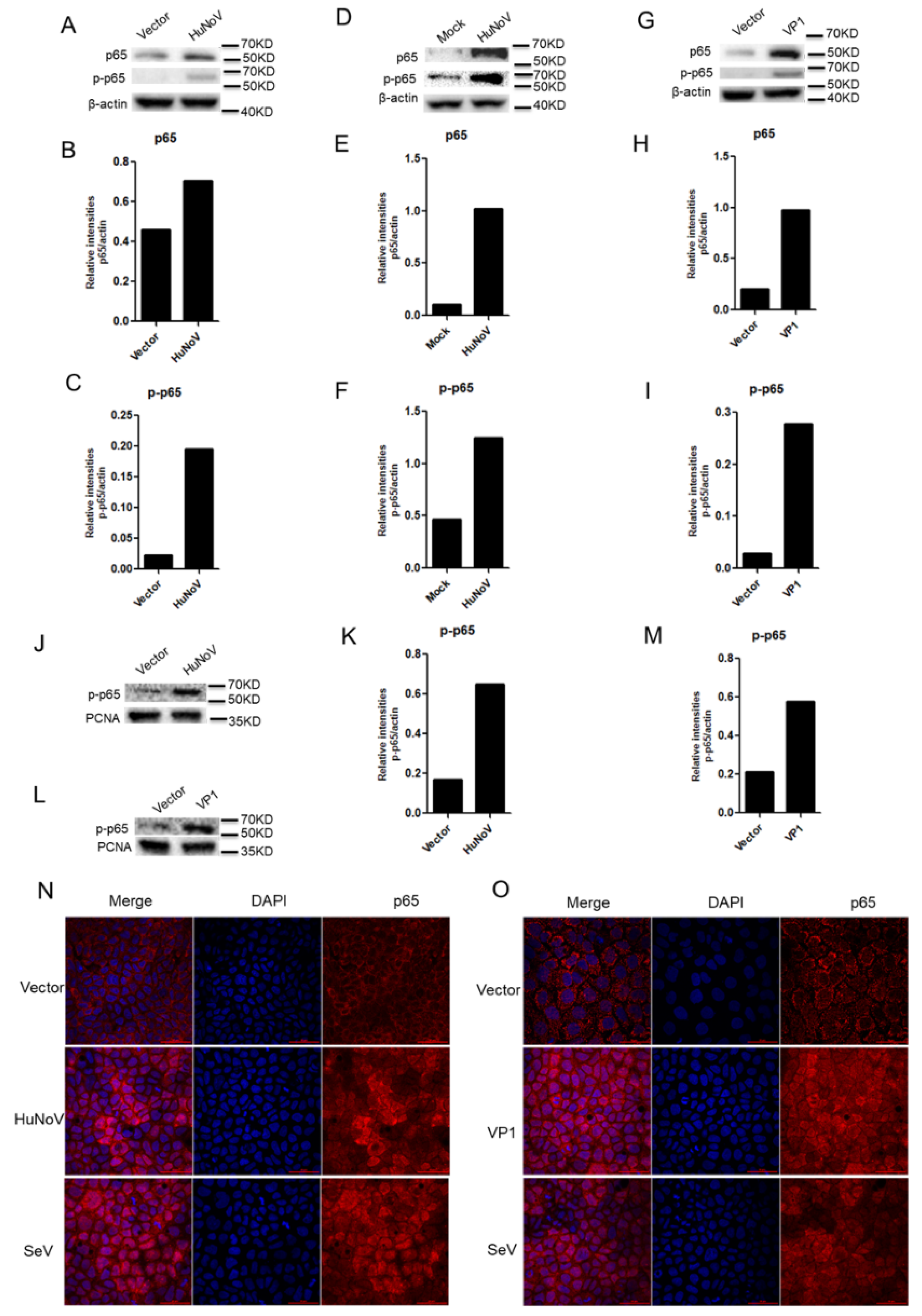
3.5. HuNoV Induces the Expression, Phosphorylation and Nuclear Translocation of p65
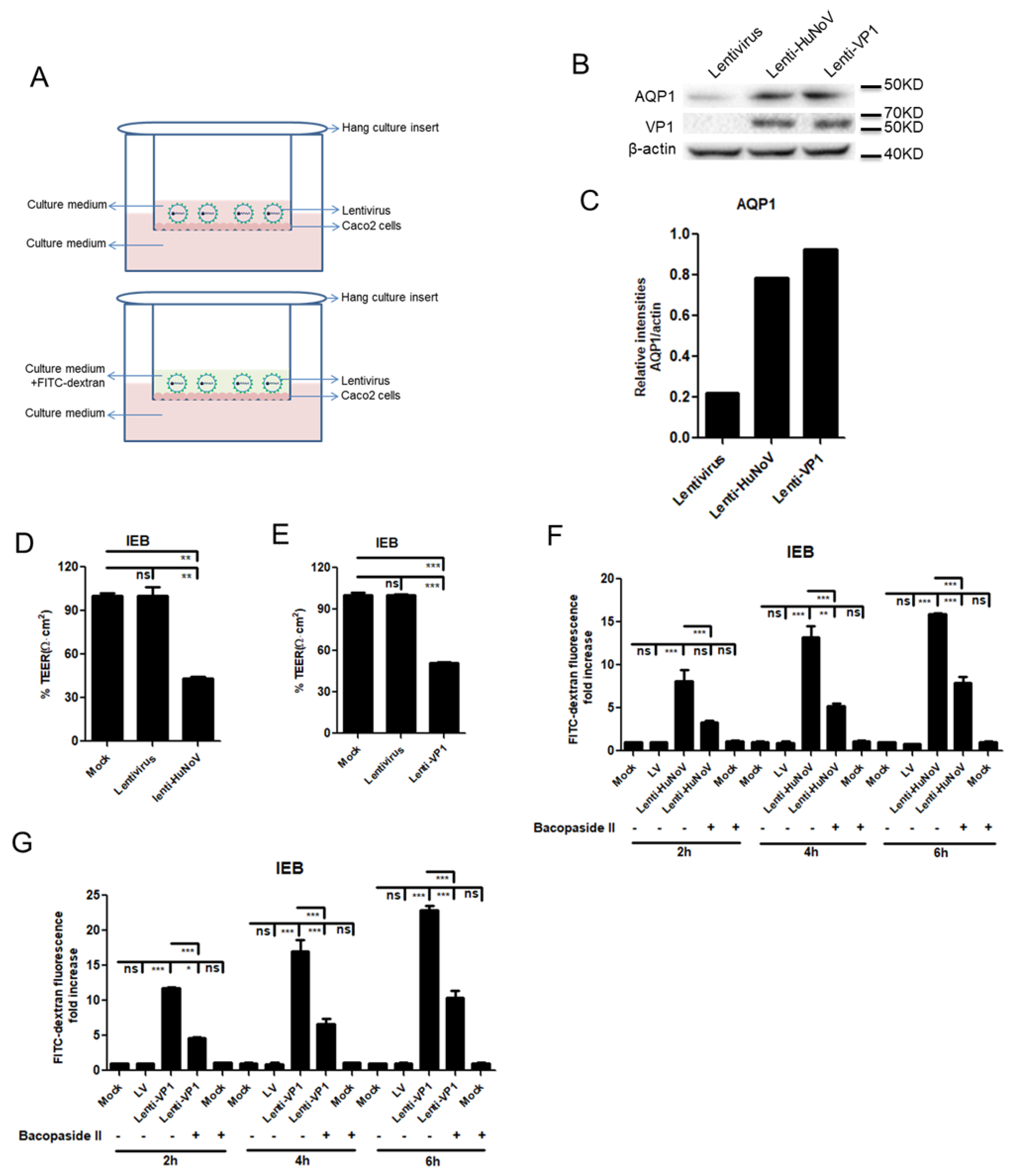
3.6. HuNoV Increases the Permeability of Intestinal Epithelial Barrier
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, R.; Neill, J.D.; Noel, J.S.; Hutson, A.M.; Glass, R.I.; Estes, M.K.; Prasad, B.V. Inter- and intragenus structural variations in caliciviruses and their functional implications. J. Virol. 2004, 78, 6469–6479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ao, Y.; Wang, J.; Ling, H.; He, Y.; Dong, X.; Wang, X.; Peng, J.; Zhang, H.; Jin, M.; Duan, Z. Norovirus GII.P16/GII.2-Associated Gastroenteritis, China, 2016. Emerg. Infect. Dis. 2017, 23, 1172–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Kroneman, A.; Vega, E.; Vennema, H.; Vinje, J.; White, P.A.; Hansman, G.; Green, K.; Martella, V.; Katayama, K.; Koopmans, M. Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 2013, 158, 2059–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.M.; Hall, A.J.; Robinson, A.E.; Verhoef, L.; Premkumar, P.; Parashar, U.D.; Koopmans, M.; Lopman, B.A. Global prevalence of norovirus in cases of gastroenteritis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti-Ciarlet, A.; White, L.J.; Chen, R.; Prasad, B.V.; Estes, M.K. Structural requirements for the assembly of Norwalk virus-like particles. J. Virol. 2002, 76, 4044–4055. [Google Scholar] [CrossRef] [Green Version]
- De Graaf, M.; van Beek, J.; Vennema, H.; Podkolzin, A.T.; Hewitt, J.; Bucardo, F.; Templeton, K.; Mans, J.; Nordgren, J.; Reuter, G.; et al. Emergence of a novel GII.17 norovirus-End of the GII.4 era? EuroSurveill 2015, 20, 21178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Ai, J.; Jin, M.; Jiang, C.; Zhang, J.; Shi, C.; Lin, Q.; Yuan, Z.; Qi, X.; Bao, C.; et al. Emergence of a new GII.17 norovirus variant in patients with acute gastroenteritis in Jiangsu, China, September 2014 to March 2015. EuroSurveill 2015, 20, 21157. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Sun, L.; Fang, L.; Yang, F.; Mo, Y.; Lao, J.; Zheng, H.; Tan, X.; Lin, H.; Rutherford, S.; et al. Gastroenteritis Outbreaks Caused by Norovirus GII.17, Guangdong Province, China, 2014-2015. Emerg. Infect. Dis. 2015, 21, 1240–1242. [Google Scholar] [CrossRef]
- Kirk, M.D.; Pires, S.M.; Black, R.E.; Caipo, M.; Crump, J.A.; Devleesschauwer, B.; Dopfer, D.; Fazil, A.; Fischer-Walker, C.L.; Hald, T.; et al. Correction: World Health Organization Estimates of the Global and Regional Disease Burden of 22 Foodborne Bacterial, Protozoal, and Viral Diseases, 2010: A Data Synthesis. PLoS Med. 2015, 12, e1001940. [Google Scholar] [CrossRef]
- Yen, C.; Hall, A.J. Editorial Commentary: Challenges to Estimating Norovirus Disease Burden. J. Pediatric Infect. Dis. Soc. 2013, 2, 61–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karst, S.M.; Wobus, C.E.; Goodfellow, I.G.; Green, K.Y.; Virgin, H.W. Advances in norovirus biology. Cell Host Microb. 2014, 15, 668–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, L.; Cina, M.; Egli-Gany, D.; Goutaki, M.; Halbeisen, F.S.; Lohrer, G.R.; Ali, H.; Scott, P.; Low, N. Prevalence of Mycoplasma genitalium in different population groups: Systematic review and meta-analysis. Sex. Trans. Inf. 2018, 94, 254–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, H.L.; Neill, F.H.; Estes, M.K.; Munoz, F.M.; Cameron, A.; DuPont, H.L.; Atmar, R.L. Noroviruses: The Most Common Pediatric Viral Enteric Pathogen at a Large University Hospital After Introduction of Rotavirus Vaccination. J. Pediatric Infect. Dis. Soc. 2013, 2, 57–60. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.J.; Lopman, B.A.; Payne, D.C.; Patel, M.M.; Gastanaduy, P.A.; Vinje, J.; Parashar, U.D. Norovirus disease in the United States. Emerg. Infect. Dis. 2013, 19, 1198–1205. [Google Scholar] [CrossRef]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef]
- Costantini, V.; Morantz, E.K.; Browne, H.; Ettayebi, K.; Zeng, X.L.; Atmar, R.L.; Estes, M.K.; Vinje, J. Human Norovirus Replication in Human Intestinal Enteroids as Model to Evaluate Virus Inactivation. Emerg. Infect. Dis. 2018, 24, 1453–1464. [Google Scholar] [CrossRef] [Green Version]
- Alvarado, G.; Ettayebi, K.; Atmar, R.L.; Bombardi, R.G.; Kose, N.; Estes, M.K.; Crowe, J.E., Jr. Human Monoclonal Antibodies That Neutralize Pandemic GII.4 Noroviruses. Gastroenterology 2018, 155, 1898–1907. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.L.F.; Rudan, I.; Liu, L.; Nair, H.; Theodoratou, E.; Bhutta, Z.A.; O’Brien, K.L.; Campbell, H.; Black, R.E. Global burden of childhood pneumonia and diarrhoea. Lancet 2013, 381, 1405–1416. [Google Scholar] [CrossRef]
- Sakai, H.; Sagara, A.; Matsumoto, K.; Hasegawa, S.; Sato, K.; Nishizaki, M.; Narita, M. 5-Fluorouracil Induces Diarrhea with Changes in the Expression of Inflammatory Cytokines and Aquaporins in Mouse Intestines. PLoS ONE 2013, 8, e54788. [Google Scholar] [CrossRef]
- Guttman, J.A.; Samji, F.N.; Li, Y.; Deng, W.; Lin, A.; Finlay, B.B. Aquaporins contribute to diarrhoea caused by attaching and effacing bacterial pathogens. Cell. Microbiol. 2007, 9, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Ikarashi, N.; Kon, R.; Sugiyama, K. Aquaporins in the Colon as a New Therapeutic Target in Diarrhea and Constipation. Int. J. Mol. Sci. 2016, 17, 1172. [Google Scholar] [CrossRef] [PubMed]
- Ikarashi, N.; Kon, R.; Iizasa, T.; Suzuki, N.; Hiruma, R.; Suenaga, K.; Toda, T.; Ishii, M.; Hoshino, M.; Ochiai, W.; et al. Inhibition of aquaporin-3 water channel in the colon induces diarrhea. Biol. Pharm. Bull. 2012, 35, 957–962. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, K.; Morishita, Y.; Tanaka, Y. The Evolutionary Aspects of Aquaporin Family. In Aquaporins; Yang, B., Ed.; Springer: Dordrecht, The Netherlands, 2017; pp. 35–50. [Google Scholar] [CrossRef]
- Kourghi, M.; Pei, J.V.; De Ieso, M.L.; Nourmohammadi, S.; Chow, P.H.; Yool, A.J. Fundamental structural and functional properties of Aquaporin ion channels found across the kingdoms of life. Clin. Exp. Pharmacol. Physiol. 2018, 45, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Verkman, A.S.; Mitra, A.K. Structure and function of aquaporin water channels. Am. J. Physiol. Renal 2000, 278, F13–F28. [Google Scholar] [CrossRef]
- Ricanek, P.; Lunde, L.K.; Frye, S.A.; Støen, M.; Nygård, S.; Morth, J.P.; Rydning, A.; Vatn, M.H.; Amiry-Moghaddam, M.; Tønjum, T. Reduced expression of aquaporins in human intestinal mucosa in early stage inflammatory bowel disease. Clin. Exp. Gastroenterol. 2015, 8, 49–67. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Deng, X.; Guan, X.; Geng, L.; Fu, M.; Zhang, B.; Chen, R.; Hu, H.; Hu, K.; Zhang, D.; et al. Herpes Simplex Virus Type 2 Infection-Induced Expression of CXCR3 Ligands Promotes CD4(+) T Cell Migration and Is Regulated by the Viral Immediate-Early Protein ICP4. Front. Immunol. 2018, 9, 2932. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Fu, M.; Li, M.; Hu, H.; Gong, S.; Hu, Q. Herpes Simplex Virus Type 2 Inhibits Type I IFN Signaling Mediated by the Novel E3 Ubiquitin Protein Ligase Activity of Viral Protein ICP22. J. Immunol. 2020, 205, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Bijvelds, M.; Dang, W.; Xu, L.; van der Eijk, A.A.; Knipping, K.; Tuysuz, N.; Dekkers, J.F.; Wang, Y.; de Jonge, J.; et al. Modeling rotavirus infection and antiviral therapy using primary intestinal organoids. Antiviral Res. 2015, 123, 120–131. [Google Scholar] [CrossRef]
- Zou, W.Y.; Blutt, S.E.; Crawford, S.E.; Ettayebi, K.; Zeng, X.L.; Saxena, K.; Ramani, S.; Karandikar, U.C.; Zachos, N.C.; Estes, M.K. Human Intestinal Enteroids: New Models to Study Gastrointestinal Virus Infections. Methods Mol. Biol. 2019, 1576, 229–247. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, K.; Su, W.; Zhao, Y.; Ma, X.; Qian, G.; Qu, G.; Pei, Z.; Liu, S.; Ma, H. Aquaporin-3 is down-regulated in jejunum villi epithelial cells during enterotoxigenic Escherichia coli-induced diarrhea in mice. Microb. Pathog. 2017, 107, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Kon, R.; Tsubota, Y.; Minami, M.; Kato, S.; Matsunaga, Y.; Kimura, H.; Murakami, Y.; Fujikawa, T.; Sakurai, R.; Tomimoto, R.; et al. CPT-11-Induced Delayed Diarrhea Develops via Reduced Aquaporin-3 Expression in the Colon. Int. J. Mol. Sci. 2018, 19, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Wu, Y.Z.; Yang, X.; Xiao, K.; Maimaitiming, A.; Gao, L.P.; Chen, C.; Gao, C.; Guo, Y.; Dong, X.P. Significant enhanced expressions of aquaporin-1, -4 and -9 in the brains of various prion diseases. Prion 2019, 13, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Kuramoto, H.; Kadowaki, M. Downregulation in aquaporin 4 and aquaporin 8 expression of the colon associated with the induction of allergic diarrhea in a mouse model of food allergy. Life Sci. 2007, 81, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Liu, L.; Wang, K.; Wu, H.; Wang, W.; Zhang, X.; Cui, G.; Cui, X.; Huang, J. Upregulation of aquaporin 3 expression by diterpenoids in Euphorbia pekinensis is associated with activation of the NF-kappaB signaling pathway in the co-culture system of HT-29 and RAW 264.7 cells. Biochimie 2018, 144, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Chen, C.; Bai, L.; Kong, L.; Luo, J. Downregulation of Aquaporin 3 Mediated the Laxative Effect in the Rat Colon by a Purified Resin Glycoside Fraction from Pharbitis Semen. Evid. Based Compl. Alternat Med. 2019, 2019, 9406342. [Google Scholar] [CrossRef] [Green Version]
- Dang, W.; Xu, L.; Ma, B.; Chen, S.; Yin, Y.; Chang, K.O.; Peppelenbosch, M.P.; Pan, Q. Nitazoxanide Inhibits Human Norovirus Replication and Synergizes with Ribavirin by Activation of Cellular Antiviral Response. Antimicrob. Agents Chemother. 2018, 62, e00707-18. [Google Scholar] [CrossRef] [Green Version]
- McFadden, N.; Bailey, D.; Carrara, G.; Benson, A.; Chaudhry, Y.; Shortland, A.; Heeney, J.; Yarovinsky, F.; Simmonds, P.; Macdonald, A.; et al. Norovirus regulation of the innate immune response and apoptosis occurs via the product of the alternative open reading frame 4. PLoS Pathog. 2011, 7, e1002413. [Google Scholar] [CrossRef] [Green Version]
- Lingemann, M.; Taube, S. Open Sesame: New Keys to Unlocking the Gate to Norovirus Infection. Cell Host Microb. 2018, 24, 463–465. [Google Scholar] [CrossRef] [Green Version]
- Bouziat, R.; Biering, S.B.; Kouame, E.; Sangani, K.A.; Kang, S.; Ernest, J.D.; Varma, M.; Brown, J.J.; Urbanek, K.; Dermody, T.S.; et al. Murine Norovirus Infection Induces TH1 Inflammatory Responses to Dietary Antigens. Cell Host Microb. 2018, 24, 677–688.e675. [Google Scholar] [CrossRef] [Green Version]
- Van Winkle, J.A.; Robinson, B.A.; Peters, A.M.; Li, L.; Nouboussi, R.V.; Mack, M.; Nice, T.J. Persistence of Systemic Murine Norovirus Is Maintained by Inflammatory Recruitment of Susceptible Myeloid Cells. Cell Host Microb. 2018, 24, 665–676.e664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, H.; Sorgeloos, F.; Sarvestani, S.T.; Martens, L.; Saeys, Y.; Mackenzie, J.M.; Lamkanfi, M.; van Loo, G.; Goodfellow, I.; Wullaert, A. Nlrp3 inflammasome activation and Gasdermin D-driven pyroptosis are immunopathogenic upon gastrointestinal norovirus infection. PLoS Pathog. 2019, 15, e1007709. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Labor. Automat. 2015, 20, 107–126. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.C.; Weidmann, M.; Albalat, A.; Carrick, E.; Morro, B.; MacKenzie, S. Polarized Trout Epithelial Cells Regulate Transepithelial Electrical Resistance, Gene Expression, and the Phosphoproteome in Response to Viral Infection. Front. Immunol. 2020, 11, 1809. [Google Scholar] [CrossRef]
- Becker, H.E.F.; Jamin, C.; Bervoets, L.; Boleij, A.; Xu, P.; Pierik, M.J.; Stassen, F.R.M.; Savelkoul, P.H.M.; Penders, J.; Jonkers, D. Higher Prevalence of Bacteroides fragilis in Crohn’s Disease Exacerbations and Strain-Dependent Increase of Epithelial Resistance. Front. Microbiol. 2021, 12, 598232. [Google Scholar] [CrossRef]
- Zhang, J.; Penny, J.; Lu, J.R. Development of a novel in vitro 3D intestinal model for permeability evaluations. Int. J. Food Sci. Nutr. 2020, 71, 549–562. [Google Scholar] [CrossRef]
- Bjorkman, E.; Casselbrant, A.; Lundberg, S.; Fandriks, L. In vitro assessment of epithelial electrical resistance in human esophageal and jejunal mucosae and in Caco-2 cell layers. Scand. J. Gastroenterol. 2012, 47, 1321–1333. [Google Scholar] [CrossRef]
- Araki, Y.; Sugihara, H.; Hattori, T. In vitro effects of dextran sulfate sodium on a Caco-2 cell line and plausible mechanisms for dextran sulfate sodium-induced colitis. Oncol. Rep. 2006, 16, 1357–1362. [Google Scholar] [CrossRef]
- Truse, R.; Nolten, I.; Schulz, J.; Herminghaus, A.; Holtmanns, T.; Gordes, L.; Raupach, A.; Bauer, I.; Picker, O.; Vollmer, C. Topical Melatonin Improves Gastric Microcirculatory Oxygenation During Hemorrhagic Shock in Dogs but Does Not Alter Barrier Integrity of Caco-2 Monolayers. Front. Med. 2020, 7, 510. [Google Scholar] [CrossRef] [PubMed]
- Glass, R.I.; Parashar, U.D.; Estes, M.K. Norovirus gastroenteritis. N. Engl. J. Med. 2009, 361, 1776–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiume, G.; Vecchio, E.; De Laurentiis, A.; Trimboli, F.; Palmieri, C.; Pisano, A.; Falcone, C.; Pontoriero, M.; Rossi, A.; Scialdone, A.; et al. Human immunodeficiency virus-1 Tat activates NF-κB via physical interaction with IκB-α and p65. Nucl. Acids Res. 2012, 40, 3548–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zhao, X.; Zhang, Z.; Guo, J.; Guan, L.; Li, J.; Mi, M.; Huang, Y.; Tong, D. Differentially expressed non-coding RNAs induced by transmissible gastroenteritis virus potentially regulate inflammation and NF-κB pathway in porcine intestinal epithelial cell line. BMC Genomics 2018, 19, 747. [Google Scholar] [CrossRef] [Green Version]
- Sumner, R.P.; Maluquer de Motes, C.; Veyer, D.L.; Smith, G.L. Vaccinia virus inhibits NF-κB-dependent gene expression downstream of p65 translocation. J. Virol. 2014, 88, 3092–3102. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Chen, Z.; Li, Y.; Zhao, Z.; He, W.; Zohaib, A.; Song, Y.; Deng, C.; Zhang, B.; Chen, H.; et al. Japanese Encephalitis Virus NS5 Inhibits Type I Interferon (IFN) Production by Blocking the Nuclear Translocation of IFN Regulatory Factor 3 and NF-κB. J. Virol. 2017, 91, e00039-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Carlin, C.R. Adenovirus early region 3 RIDα protein limits NFκB signaling through stress-activated EGF receptors. PLoS Pathog. 2019, 15, e1008017. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Zhang, B.; Chen, R.; Li, M.; Zheng, Z.; Xu, W.; Zhang, Y.; Gong, S.; Hu, Q. Human Norovirus Induces Aquaporin 1 Production by Activating NF-κB Signaling Pathway. Viruses 2022, 14, 842. https://doi.org/10.3390/v14040842
Zhang M, Zhang B, Chen R, Li M, Zheng Z, Xu W, Zhang Y, Gong S, Hu Q. Human Norovirus Induces Aquaporin 1 Production by Activating NF-κB Signaling Pathway. Viruses. 2022; 14(4):842. https://doi.org/10.3390/v14040842
Chicago/Turabian StyleZhang, Mudan, Binman Zhang, Rui Chen, Miaomiao Li, Zifeng Zheng, Wanfu Xu, Yifan Zhang, Sitang Gong, and Qinxue Hu. 2022. "Human Norovirus Induces Aquaporin 1 Production by Activating NF-κB Signaling Pathway" Viruses 14, no. 4: 842. https://doi.org/10.3390/v14040842