
Identification of Transcription Factors Regulating SARS-CoV-2 Tropism Factor Expression by Inferring Cell-Type-Specific Transcriptional Regulatory Networks in Human Lungs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
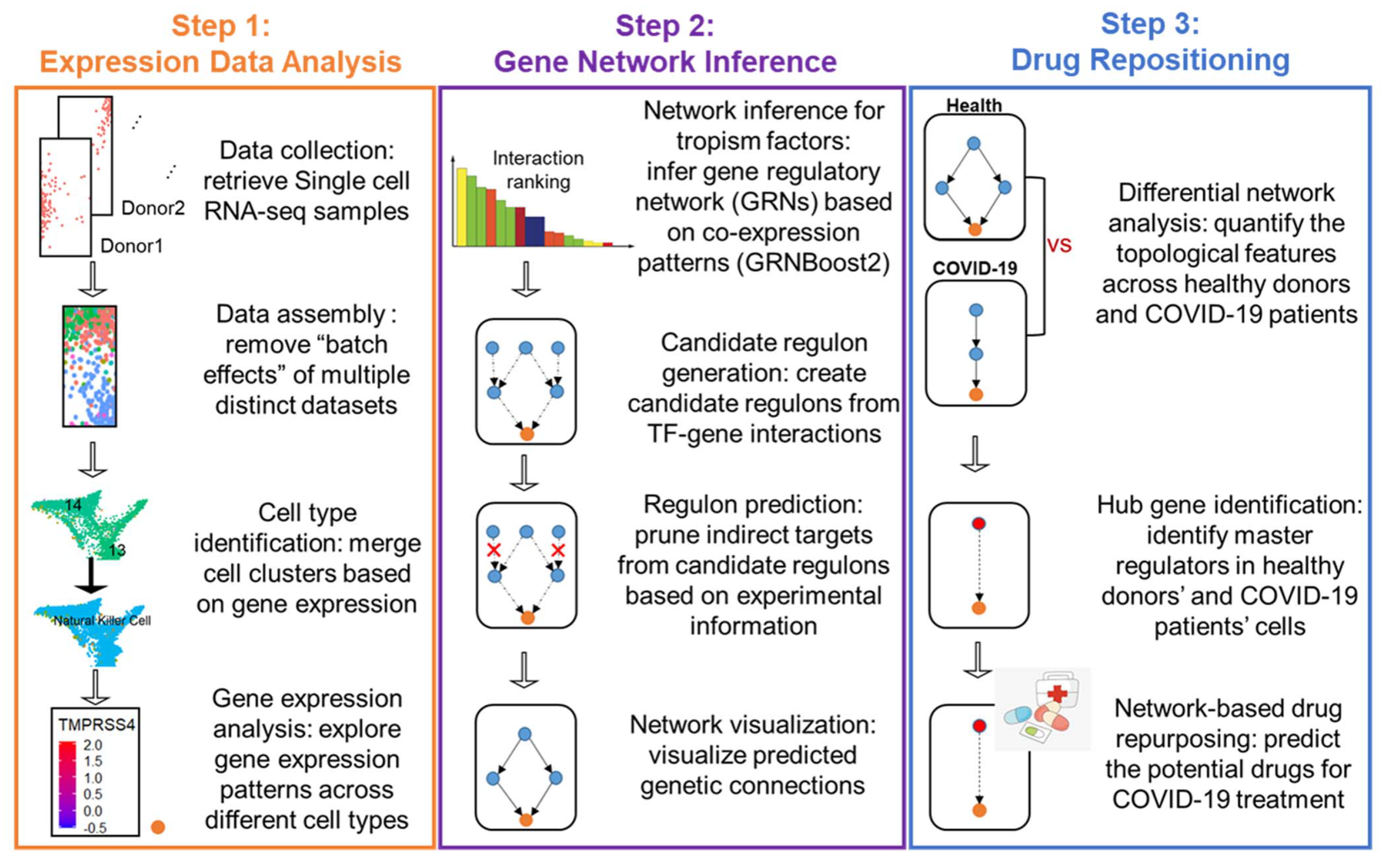
2. Materials and Methods
2.1. Acquisition and Procession of Public Datasets
2.2. Assembly of Multiple Distinct scRNA-Seq Datasets
2.3. Unsupervised Dimensionality Reduction and Clustering
2.4. Marker Gene Selection and the Classification of Cell Types
2.5. Module Generation and Direct TF–Target Regulon Identification
2.6. Defining the Regulatory Direction of Each TF–Target Regulon
2.7. Network Organization and Visualization
2.8. Differential Expression Analysis
2.9. Differential Network Analysis
2.10. Hybrid Centrality Measures
2.11. Mapping Drug Targets
3. Results
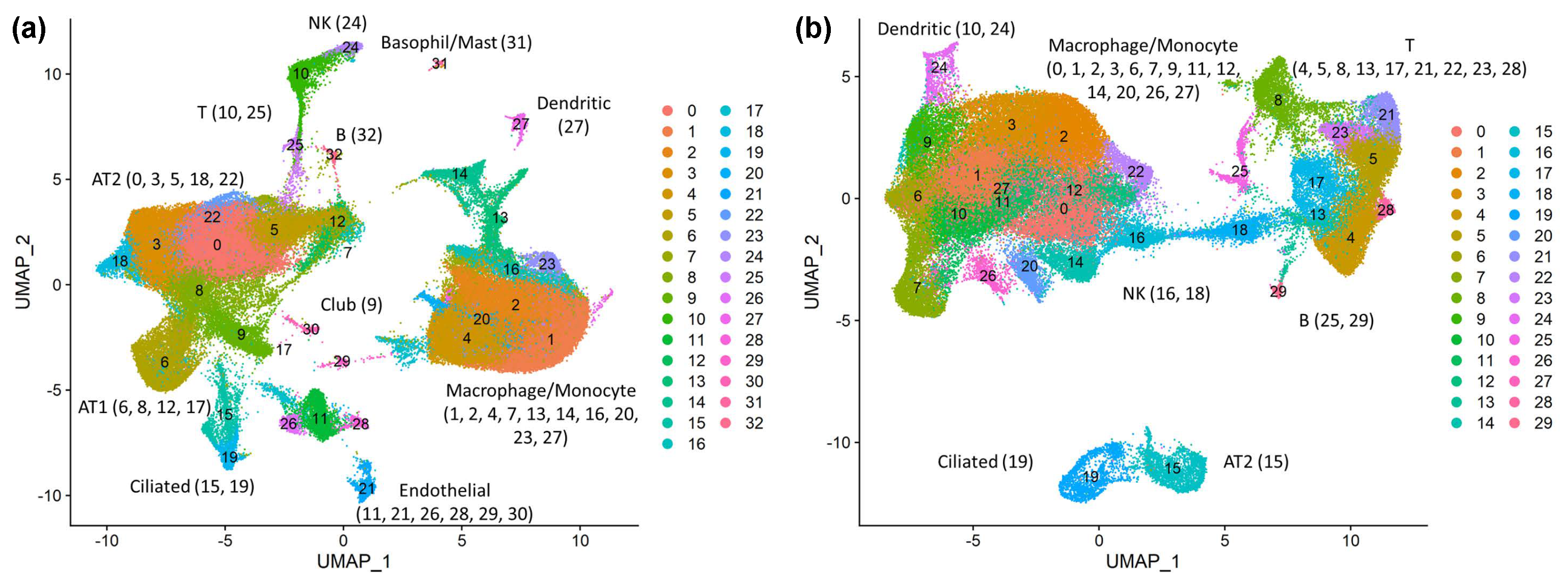
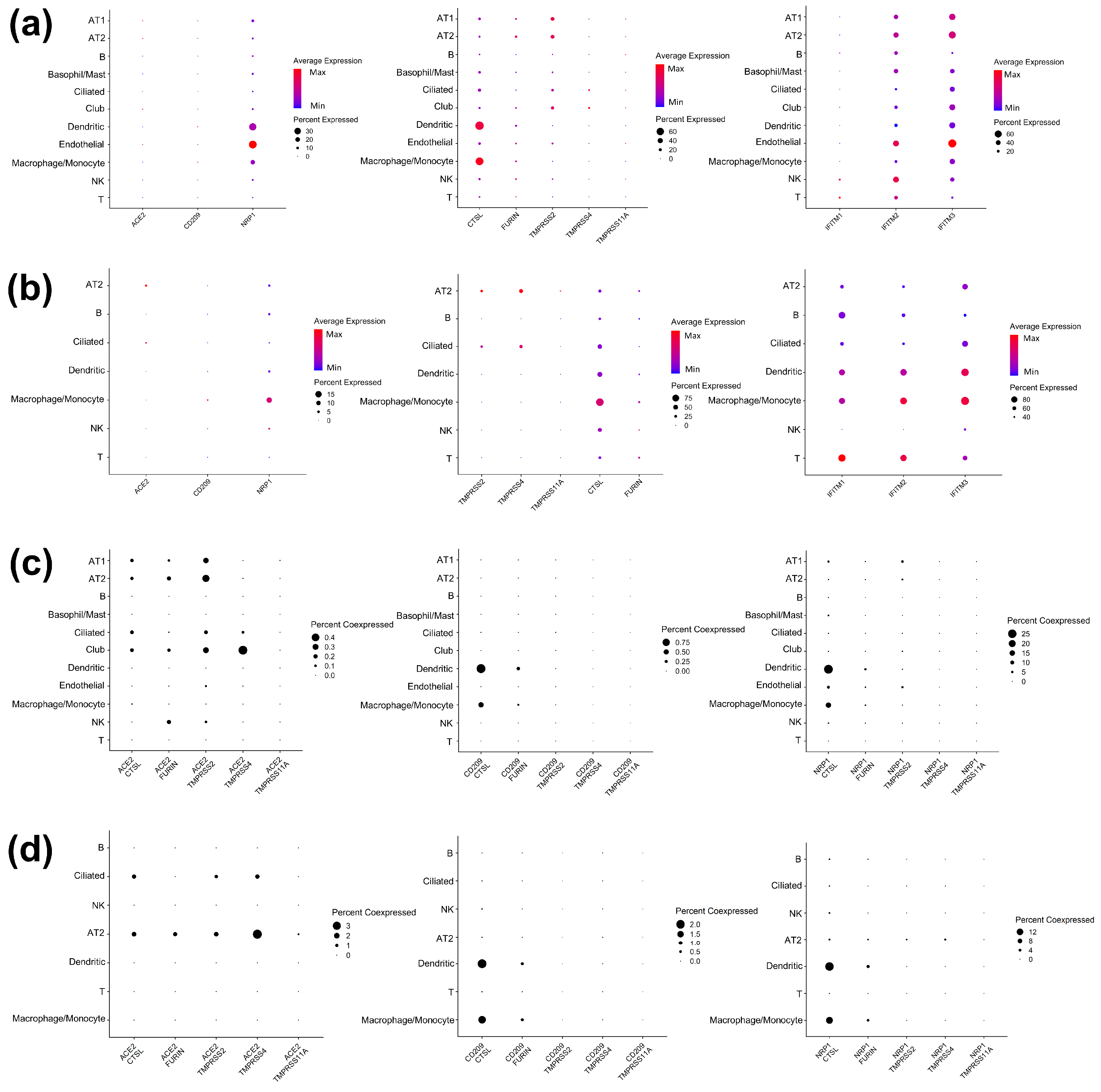
3.1. Expression Landscape of the SARS-CoV-2 Tropism Factors Are Largely Different in Each Cell Type
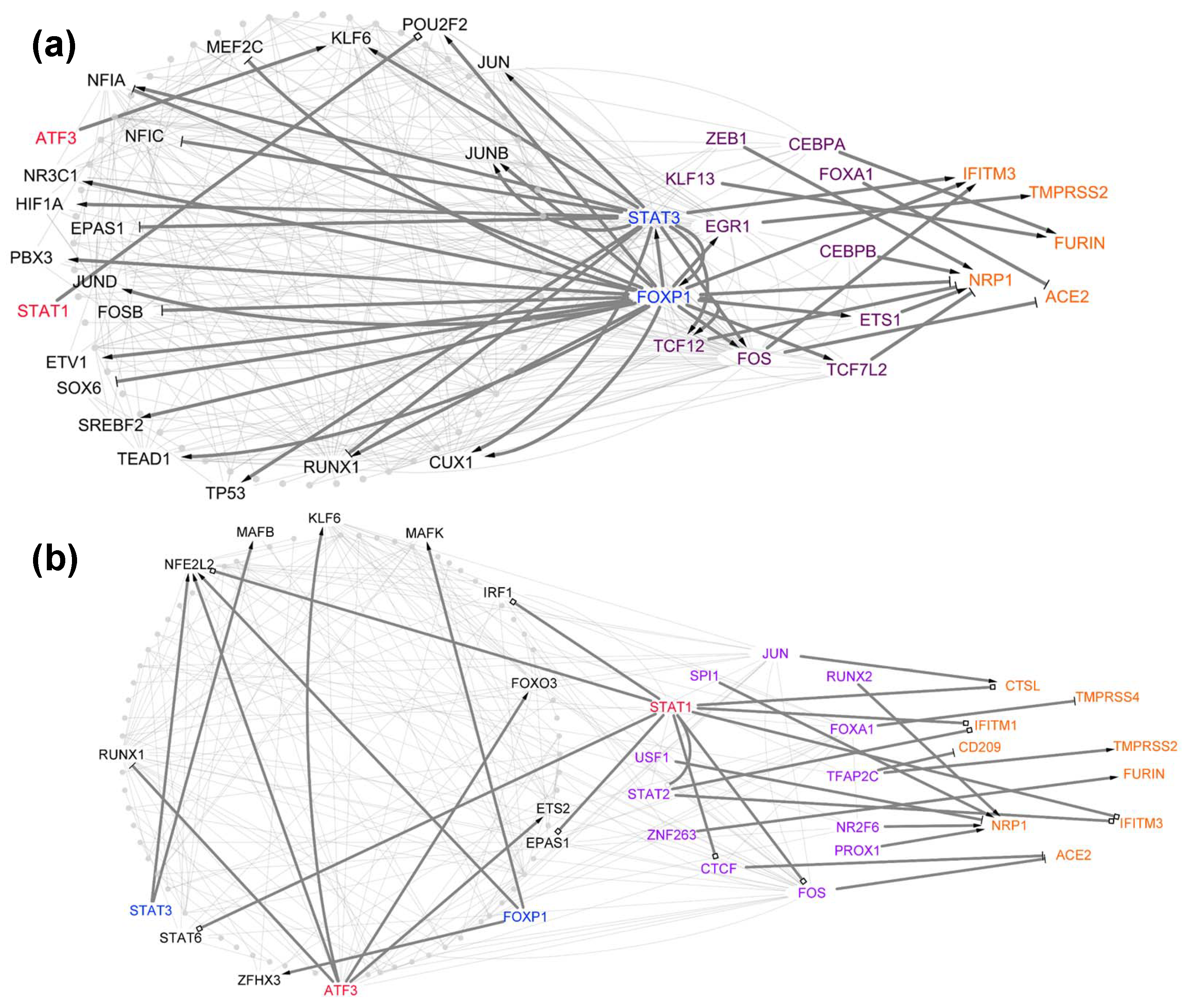
3.2. Cell-Type-Specific Trns Affecting Tropism Factors
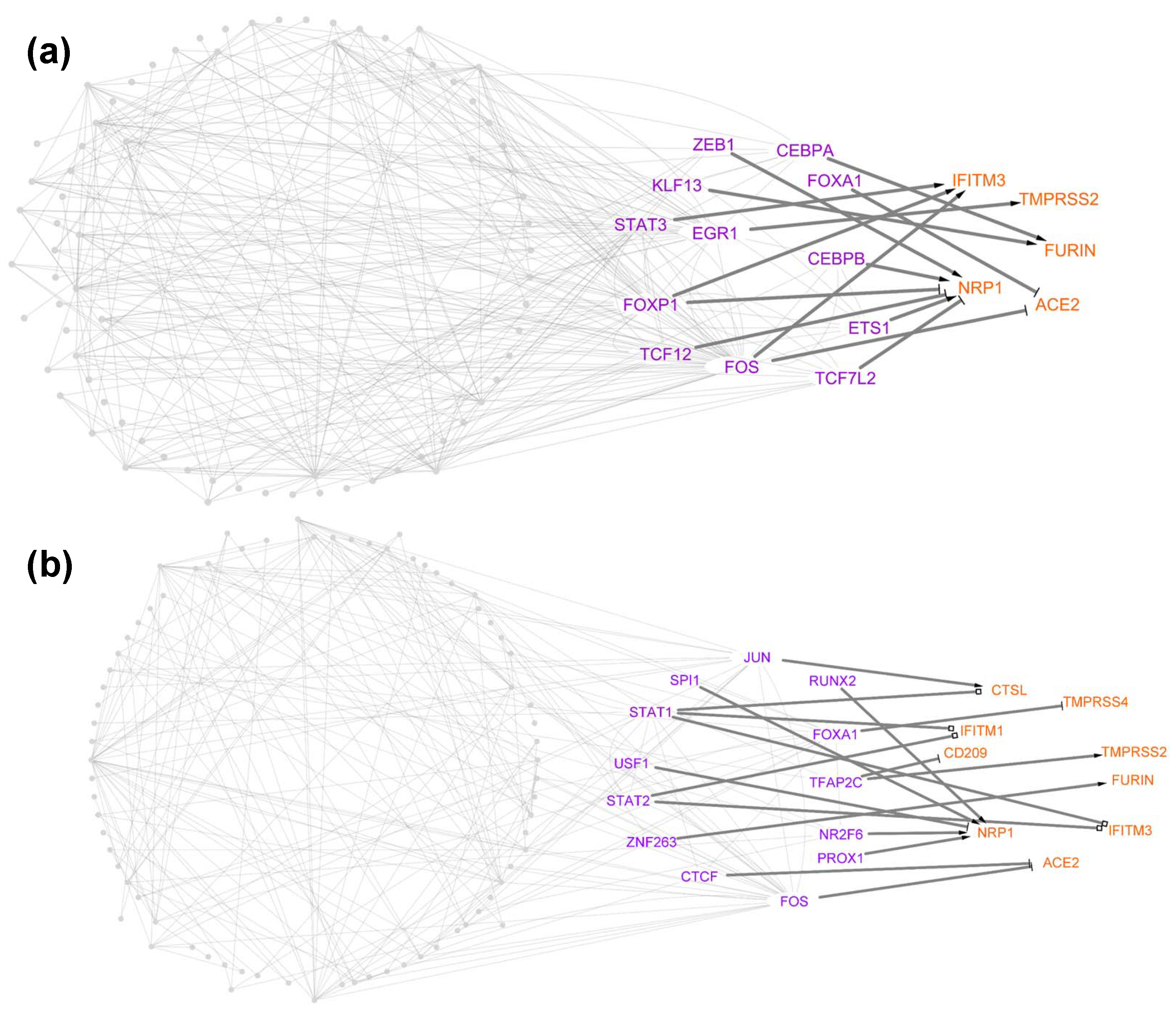
3.3. Identification of TFs Directly Regulating Tropism Factors within Each Cell Type
3.4. Centrality Analysis for Predicting TFs That Are Important in Host Cell Tropism
4. Discussion
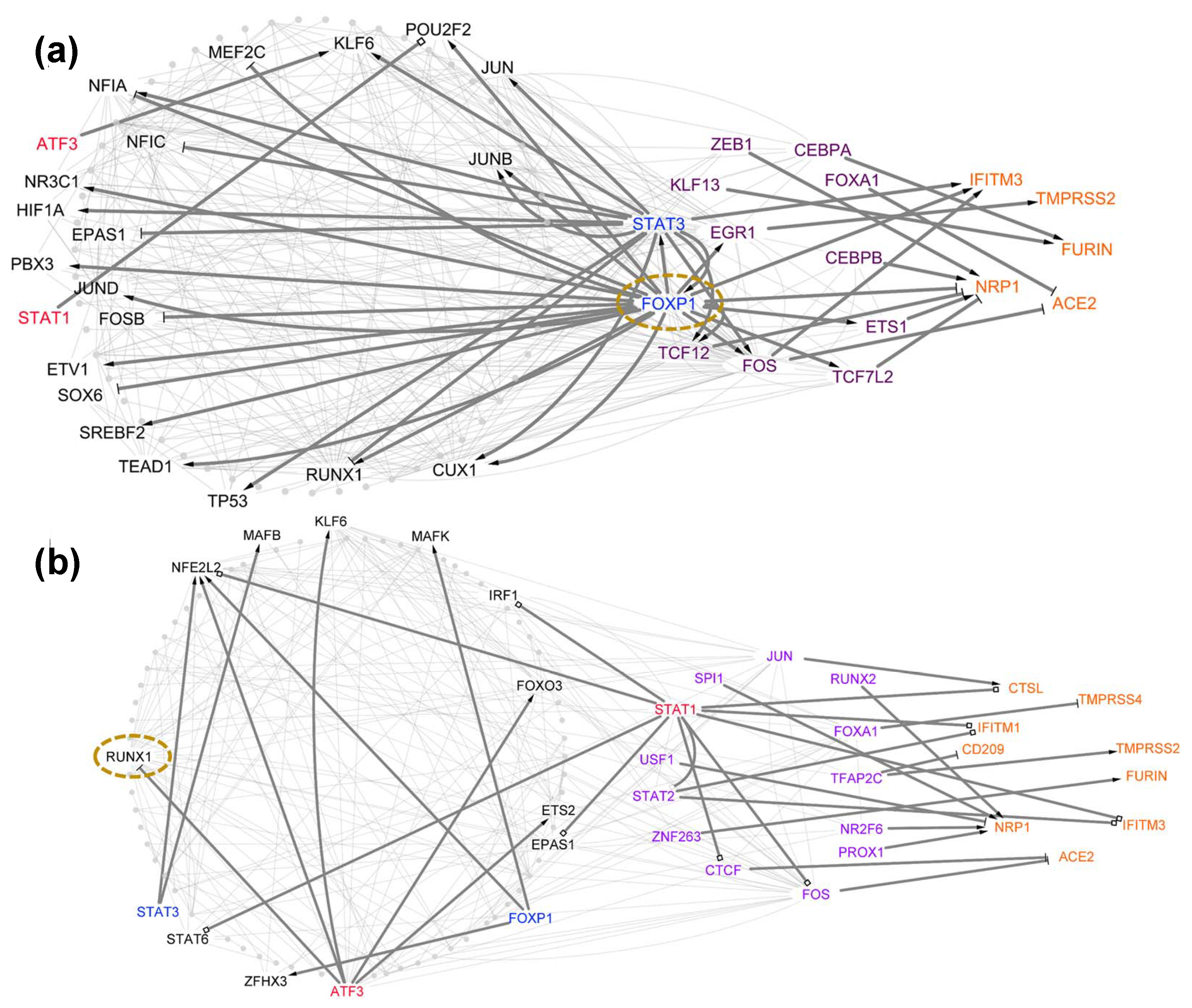
4.1. Case Study 1: Stat1 Has a Different Regulatory Role for Host Cell Tropism Factors in Healthy vs. SARS-CoV-2-Infected Donors
4.2. Case Study 2: Differential Centrality Analysis Can Be Used to Identify Important TFs Whose Expression Is Not Differentially Changed between Healthy and SARS-CoV-2-Infected Donors
4.3. Case Study 3: Hub Tfs Identified by Network Centrality Analysis Could Be Used as Targets for Drug Repositioning to Prevent SARS-CoV-2 Infection and Transmission
4.4. Limitations of the Present Approach
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hopkins, H. COVID-19 Case Tracker. Follow Global Cases and Trends. 2020. Available online: https://coronavirus.jhu.edu/map.html (accessed on 2 March 2022).
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Bradley, B.T.; Maioli, H.; Johnston, R.; Chaudhry, I.; Fink, S.L.; Xu, H.; Najafian, B.; Deutsch, G.; Lacy, J.M.; Williams, T.; et al. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: A case series. Lancet 2020, 396, 320–332. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, N. Generalized anxiety disorder, depressive symptoms and sleep quality during COVID-19 outbreak in China: A web-based cross-sectional survey. Psychiatry Res. 2020, 288, 112954. [Google Scholar] [CrossRef]
- Xu, J.; Xu, X.; Jiang, L.; Dua, K.; Hansbro, P.M.; Liu, G. SARS-CoV-2 induces transcriptional signatures in human lung epithelial cells that promote lung fibrosis. Respir. Res. 2020, 21, 1–12. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Singh, M.; Bansal, V.; Feschotte, C. A single-cell RNA expression map of human coronavirus entry factors. Cell Rep. 2020, 32, 108175. [Google Scholar] [CrossRef]
- Vankadari, N.; Wilce, J.A. Emerging COVID-19 coronavirus: Glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg. Microbes Infect. 2020, 9, 601–604. [Google Scholar] [CrossRef]
- Wang, P.G.; Tang, D.J.; Hua, Z.; Wang, Z.; An, J. Sunitinib reduces the infection of SARS-CoV, MERS-CoV and SARS-CoV-2 partially by inhibiting AP2M1 phosphorylation. Cell Discov. 2020, 6, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Amraei, R.; Yin, W.; Napoleon, M.A.; Suder, E.L.; Berrigan, J.; Zhao, Q.; Olejnik, J.; Chandler, K.B.; Xia, C.; Feldman, J.; et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2. ACS Cent. Sci. 2021, 7, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, C.R.; Di Fazio, A.; Nadvi, N.A.; Patel, K.; Xiang, M.S.W.; Zhang, H.E.; Deshpande, C.; Low, J.K.; Wang, X.T.; Chen, Y.; et al. A novel purification procedure for active recombinant human DPP4 and the inability of DPP4 to bind SARS-CoV-2. Molecules 2020, 25, 5392. [Google Scholar] [CrossRef] [PubMed]
- Tsou, E.P.S.; Sule, G.; Rubio, M.G.; Amin, M.A.; Zuo, Y.; Knight, J.; Kanthi, Y.; Fox, D. Identification of CD13 as a Potential Cause for SARS-CoV-2-triggered Hyperinflammation and Thrombosis. Arthritis Rheumatol. 2020, 72. [Google Scholar]
- Yang, L.; Han, Y.; Nilsson-Payant, B.E.; Gupta, V.; Wang, P.; Duan, X.; Tang, X.; Zhu, J.; Zhao, Z.; Jaffré, F.; et al. A human pluripotent stem cell-based platform to study SARS-CoV-2 tropism and model virus infection in human cells and organoids. Cell Stem Cell 2020, 27, 125–136. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 2020, 78, 779–784. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; Zhang, L.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef]
- Zang, R.; Castro, M.F.G.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Y.; Zhang, S.; Wang, Z.; Sun, S.; Liu, M.; Chen, Y.; Dong, N.; Wu, Q. Intracellular autoactivation of TMPRSS11A, an airway epithelial transmembrane serine protease. J. Biol. Chem. 2020, 295, 12686–12696. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.C.; Zhong, G.; Huang, I.C.; Farzan, M. IFITM-family proteins: The cell’s first line of antiviral defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozzo, C.P.; Nchioua, R.; Volcic, M.; Wettstein, L.; Weil, T.; Krüger, J.; Heller, S.; Conzelmann, C.; Müller, J.; Gross, R.; et al. IFITM proteins promote SARS-CoV-2 infection and are targets for virus inhibition in vitro. Nat. Commun. 2021, 12, 4584. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Kenney, A.D.; Kudryashova, E.; Zani, A.; Zhang, L.; Lai, K.K.; Hall-Stoodley, L.; Robinson, R.T.; Kudryashov, D.S.; Compton, A.A.; et al. Opposing activities of IFITM proteins in SARS-CoV-2 infection. EMBO J. 2021, 40, e106501. [Google Scholar] [CrossRef]
- Barabási, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.X.; Thomas, C.E.; Brunak, S. Network biology concepts in complex disease comorbidities. Nat. Rev. Genet. 2016, 17, 615–629. [Google Scholar] [CrossRef]
- Cagno, V. SARS-CoV-2 cellular tropism. Lancet Microbe 2020, 1, e2. [Google Scholar] [CrossRef]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon III, K.H.; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 2020, 182, 429–446. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef]
- Qi, F.; Qian, S.; Zhang, S.; Zhang, Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem. Biophys. Res. Commun. 2020, 526, 135–140. [Google Scholar] [CrossRef]
- Murgolo, N.; Therien, A.G.; Howell, B.; Klein, D.; Koeplinger, K.; Lieberman, L.A.; Adam, G.C.; Flynn, J.; McKenna, P.; Swaminathan, G.; et al. SARS-CoV-2 tropism, entry, replication, and propagation: Considerations for drug discovery and development. PLoS Pathog. 2021, 17, e1009225. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, A. From ‘differential expression’ to ‘differential networking’—Identification of dysfunctional regulatory networks in diseases. Trends Genet. 2010, 26, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Del Sol, A.; Balling, R.; Hood, L.; Galas, D. Diseases as network perturbations. Curr. Opin. Biotechnol. 2010, 21, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Jalili, M.; Salehzadeh-Yazdi, A.; Gupta, S.; Wolkenhauer, O.; Yaghmaie, M.; Resendis-Antonio, O.; Alimoghaddam, K. Evolution of centrality measurements for the detection of essential proteins in biological networks. Front. Physiol. 2016, 7, 375. [Google Scholar] [CrossRef] [Green Version]
- Iacono, G.; Massoni-Badosa, R.; Heyn, H. Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biol. 2019, 20, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Resendis-Antonio, O.; Freyre-Gonzalez, J.A.; Menchaca-Mendez, R.; Gutiérrez-Ríos, R.M.; Martínez-Antonio, A.; Avila-Sanchez, C.; Collado-Vides, J. Modular analysis of the transcriptional regulatory network of E. coli. Trends Genet. 2005, 21, 16–20. [Google Scholar] [CrossRef]
- Resendis-Antonio, O.; Hernández, M.; Mora, Y.; Encarnación, S. Functional modules, structural topology, and optimal activity in metabolic networks. PLoS Comput. Biol. 2012, 8, e1002720. [Google Scholar] [CrossRef] [Green Version]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Grant, R.A.; Morales-Nebreda, L.; Markov, N.S.; Swaminathan, S.; Querrey, M.; Guzman, E.R.; Abbott, D.A.; Donnelly, H.K.; Donayre, A.; Goldberg, I.A.; et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature 2021, 590, 635–641. [Google Scholar] [CrossRef]
- Morse, C.; Tabib, T.; Sembrat, J.; Buschur, K.L.; Bittar, H.T.; Valenzi, E.; Jiang, Y.; Kass, D.J.; Gibson, K.; Chen, W.; et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 54. [Google Scholar] [CrossRef] [PubMed]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE 2 and TMPRSS 2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef] [PubMed]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck III, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single-cell data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Haghverdi, L.; Lun, A.T.; Morgan, M.D.; Marioni, J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018, 36, 421–427. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Madhulatha, T.S. An overview on clustering methods. arXiv 2012, arXiv:1205.1117. [Google Scholar] [CrossRef]
- Xu, C.; Su, Z. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics 2015, 31, 1974–1980. [Google Scholar] [CrossRef] [Green Version]
- Blondel, V.D.; Guillaume, J.L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef] [Green Version]
- Waltman, L.; Van Eck, N.J. A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B 2013, 86, 1–14. [Google Scholar] [CrossRef]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Moerman, T.; Aibar Santos, S.; Bravo González-Blas, C.; Simm, J.; Moreau, Y.; Aerts, J.; Aerts, S. GRNBoost2 and Arboreto: Efficient and scalable inference of gene regulatory networks. Bioinformatics 2019, 35, 2159–2161. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alonso, L.; Holland, C.H.; Ibrahim, M.M.; Turei, D.; Saez-Rodriguez, J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019, 29, 1363–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatake, Y.; Ko, S.B.; Sharov, A.A.; Wakabayashi, S.; Murakami, M.; Sakota, M.; Chikazawa, N.; Ookura, C.; Sato, S.; Ito, N.; et al. Generation and profiling of 2,135 human ESC lines for the systematic analyses of cell states perturbed by inducing single transcription factors. Cell Rep. 2020, 31, 107655. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Song, C.; Liu, Y.; Qian, F.; Gao, Y.; Ning, Z.; Wang, Q.; Jiang, Y.; Li, Y.; Li, M.; et al. KnockTF: A comprehensive human gene expression profile database with knockdown/knockout of transcription factors. Nucleic Acids Res. 2020, 48, D93–D100. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef]
- Van de Sande, B.; Flerin, C.; Davie, K.; De Waegeneer, M.; Hulselmans, G.; Aibar, S.; Seurinck, R.; Saelens, W.; Cannoodt, R.; Rouchon, Q.; et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nature Protoc. 2020, 15, 2247–2276. [Google Scholar] [CrossRef]
- Ding, H.; Yang, Y.; Xue, Y.; Seninge, L.; Gong, H.; Safavi, R.; Califano, A.; Stuart, J.M. Prioritizing transcriptional factors in gene regulatory networks with PageRank. Iscience 2021, 24, 102017. [Google Scholar] [CrossRef]
- Koschützki, D.; Schreiber, F. Centrality analysis methods for biological networks and their application to gene regulatory networks. Gene Regul. Syst. Biol. 2008, 2, GRSB-S702. [Google Scholar] [CrossRef]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- Skuta, C.; Popr, M.; Muller, T.; Jindrich, J.; Kahle, M.; Sedlak, D.; Svozil, D.; Bartunek, P. Probes & Drugs portal: An interactive, open data resource for chemical biology. Nat. Methods 2017, 14, 759–760. [Google Scholar] [PubMed]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Dorrity, M.W.; Saunders, L.M.; Queitsch, C.; Fields, S.; Trapnell, C. Dimensionality reduction by UMAP to visualize physical and genetic interactions. Nature Commun. 2020, 11, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, C.G.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 2020, 181, 1016–1035. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.M.; Yang, W.L.; Yang, F.Y.; Zhang, L.; Huang, W.J.; Hou, W.; Fan, C.F.; Jin, R.H.; Feng, Y.M.; Wang, Y.C.; et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 2021, 6, 1–12. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The human transcription factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, K.B.; Chodavarapu, H.; Lazartigues, E. Forkhead box transcription factors of the FOXA class are required for basal transcription of angiotensin-converting enzyme 2. J. Endocr. Soc. 2017, 1, 370–384. [Google Scholar] [CrossRef] [Green Version]
- Hennighausen, L.; Lee, H.K. Activation of the SARS-CoV-2 receptor Ace2 through JAK/STAT-dependent enhancers during pregnancy. Cell Rep. 2020, 32, 108199. [Google Scholar] [CrossRef]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020, 27, 3209–3225. [Google Scholar] [CrossRef] [PubMed]
- Hogan, R.J.; Gao, G.; Rowe, T.; Bell, P.; Flieder, D.; Paragas, J.; Kobinger, G.P.; Wivel, N.A.; Crystal, R.G.; Boyer, J.; et al. Resolution of primary severe acute respiratory syndrome-associated coronavirus infection requires Stat1. J. Virol. 2004, 78, 11416–11421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parolia, A.; Cieslik, M.; Chu, S.C.; Xiao, L.; Ouchi, T.; Zhang, Y.; Wang, X.; Vats, P.; Cao, X.; Pitchiaya, S.; et al. Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 2019, 571, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Wang, X.M.; Mannan, R.; Pitchiaya, S.; Zhang, Y.; Wotring, J.W.; Xiao, L.; Robinson, D.R.; Wu, Y.M.; Tien, J.C.Y.; et al. Targeting transcriptional regulation of SARS-CoV-2 entry factors ACE2 and TMPRSS2. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Chilakamarti, R.; Bikul, D. Regulation of early growth response-1 (Egr-1) gene expression by Stat1-independent type I interferon signaling and respiratory viruses. Comput. Math. Biophys. 2021, 1, 289–303. [Google Scholar]
- Patra, T.; Meyer, K.; Geerling, L.; Isbell, T.S.; Hoft, D.F.; Brien, J.; Pinto, A.K.; Ray, R.B.; Ray, R. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PLoS Pathog. 2020, 16, e1009128. [Google Scholar] [CrossRef] [PubMed]
- Mayi, B.S.; Leibowitz, J.A.; Woods, A.T.; Ammon, K.A.; Liu, A.E.; Raja, A. The role of Neuropilin-1 in COVID-19. PLoS Pathog. 2021, 17, e1009153. [Google Scholar] [CrossRef]
- He, R.; Leeson, A.; Andonov, A.; Li, Y.; Bastien, N.; Cao, J.; Osiowy, C.; Dobie, F.; Cutts, T.; Ballantine, M.; et al. Activation of AP-1 signal transduction pathway by SARS coronavirus nucleocapsid protein. Biochem. Biophys. Res. Commun. 2003, 311, 870–876. [Google Scholar] [CrossRef]
- Wang, H.; Li, X.; Li, T.; Zhang, S.; Wang, L.; Wu, X.; Liu, J. The genetic sequence, origin, and diagnosis of SARS-CoV-2. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1629–1635. [Google Scholar] [CrossRef]
- Bagheri, A.; Moezzi, S.M.I.; Mosaddeghi, P.; Parashkouhi, S.N.; Hoseini, S.M.F.; Badakhshan, F.; Negahdaripour, M. Interferon-inducer antivirals: Potential candidates to combat COVID-19. Int. Immunopharmacol. 2021, 91, 107245. [Google Scholar] [CrossRef]
- Chakrabarty, B.; Das, D.; Bulusu, G.; Roy, A. Network-based analysis of fatal comorbidities of COVID-19 and potential therapeutics. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.T.A.; Khalid, Z.; Zahid, H.; Yousaf, M.A.; Shakoori, A.R. A computational and bioinformatic analysis of ACE2: An elucidation of its dual role in COVID-19 pathology and finding its associated partners as potential therapeutic targets. J. Biomol. Struct. Dyn. 2020, 40, 1813–1829. [Google Scholar] [CrossRef] [PubMed]
- Mamoor, S. The Activating Transcription Factor ATF3 is Differentially Expressed and Transcriptionally Induced in Multiple Models of Coronavirus Infection. 2020. Available online: https://osf.io/dhnpm (accessed on 2 March 2022).
- Shamir, I.; Abutbul-Amitai, M.; Abbas-Egbariya, H.; Pasmanik-Chor, M.; Paret, G.; Nevo-Caspi, Y. STAT3 isoforms differentially affect ACE2 expression: A potential target for COVID-19 therapy. J. Cell. Mol. Med. 2020, 24, 12864–12868. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Yin, X.; Meng, X.; Chan, J.F.W.; Ye, Z.W.; Riva, L.; Pache, L.; Chan, C.C.Y.; Lai, P.M.; Chan, C.C.S.; et al. Clofazimine broadly inhibits coronaviruses including SARS-CoV-2. Nature 2021, 593, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Y.; Liu, Q.; Yao, Q.; Wang, X.; Zhang, H.; Chen, R.; Ren, L.; Min, J.; Deng, F.; et al. SARS-CoV-2 cell tropism and multiorgan infection. Cell Discov. 2021, 7, 1–4. [Google Scholar] [CrossRef]
- Mu, J.; Fang, Y.; Yang, Q.; Shu, T.; Wang, A.; Huang, M.; Jin, L.; Deng, F.; Qiu, Y.; Zhou, X. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov. 2020, 6, 1–4. [Google Scholar] [CrossRef]
- Myers, J.S.; von Lersner, A.K.; Robbins, C.J.; Sang, Q.X.A. Differentially expressed genes and signature pathways of human prostate cancer. PLoS ONE 2015, 10, e0145322. [Google Scholar] [CrossRef]
- Murata, M.; Yoshida, K.; Yamaguchi, T.; Matsuzaki, K. Linker phosphorylation of Smad3 promotes fibro-carcinogenesis in chronic viral hepatitis of hepatocellular carcinoma. World J. Gastroenterol. WJG 2014, 20, 15018. [Google Scholar] [CrossRef]
- Cowley, D.O.; Graves, B.J. Phosphorylation represses Ets-1 DNA binding by reinforcing autoinhibition. Genes Dev. 2000, 14, 366–376. [Google Scholar] [CrossRef]
- Nasr, R.; Guillemin, M.C.; Ferhi, O.; Soilihi, H.; Peres, L.; Berthier, C.; Rousselot, P.; Robledo-Sarmiento, M.; Lallemand-Breitenbach, V.; Gourmel, B.; et al. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat. Med. 2008, 14, 1333–1342. [Google Scholar] [CrossRef]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Hiasa, Y.; Tokumoto, Y.; Hirooka, M.; Abe, M.; Ikeda, Y.; Matsuura, B.; Chung, R.T.; Onji, M. Protein kinase R modulates c-Fos and c-Jun signaling to promote proliferation of hepatocellular carcinoma with hepatitis C virus infection. PLoS ONE 2013, 8, e67750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hock, H.; Shimamura, A. ETV6 in hematopoiesis and leukemia predisposition. In Seminars in Hematology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 54, pp. 98–104. [Google Scholar]
- Rocca, D.L.; Wilkinson, K.A.; Henley, J.M. SUMOylation of FOXP1 regulates transcriptional repression via CtBP1 to drive dendritic morphogenesis. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunz, K.; Wagner, K.; Mendler, L.; Hölper, S.; Dehne, N.; Müller, S. SUMO signaling by hypoxic inactivation of SUMO-specific isopeptidases. Cell Rep. 2016, 16, 3075–3086. [Google Scholar] [CrossRef] [Green Version]
- Illendula, A.; Gilmour, J.; Grembecka, J.; Tirumala, V.S.S.; Boulton, A.; Kuntimaddi, A.; Schmidt, C.; Wang, L.; Pulikkan, J.A.; Zong, H.; et al. Small molecule inhibitor of CBFβ-RUNX binding for RUNX transcription factor driven cancers. EBioMedicine 2016, 8, 117–131. [Google Scholar] [CrossRef] [Green Version]
- Mason, R.J. Pathogenesis of COVID-19 from a Cell Biology Perspective. Eur. Respir. J. 2020, 55, 2000607. [Google Scholar] [CrossRef] [Green Version]
- Mulay, A.; Konda, B.; Garcia, G., Jr.; Yao, C.; Beil, S.; Villalba, J.M.; Koziol, C.; Sen, C.; Purkayastha, A.; Kolls, J.K.; et al. SARS-CoV-2 infection of primary human lung epithelium for COVID-19 modeling and drug discovery. Cell Rep. 2021, 35, 109055. [Google Scholar] [CrossRef]
- An, X.; Zhang, Y.; Duan, L.; Jin, D.; Zhao, S.; Zhou, R.; Duan, Y.; Lian, F.; Tong, X. The Direct Evidence and Mechanism of Traditional Chinese Medicine Treatment of COVID-19. Biomed. Pharmacother. 2021, 137, 111267. [Google Scholar] [CrossRef]
- Zhang, L.; Li, J.; Zhou, M.; Chen, Z. Summary of 20 tracheal intubation by anesthesiologists for patients with severe COVID-19 pneumonia: Retrospective case series. J. Anesth. 2020, 34, 599–606. [Google Scholar] [CrossRef]
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR screens reveal host factors critical for SARS-CoV-2 infection. Cell 2021, 184, 76–91. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tong, H.; Chen, H.; Williams, C.M. Identification of Transcription Factors Regulating SARS-CoV-2 Tropism Factor Expression by Inferring Cell-Type-Specific Transcriptional Regulatory Networks in Human Lungs. Viruses 2022, 14, 837. https://doi.org/10.3390/v14040837
Tong H, Chen H, Williams CM. Identification of Transcription Factors Regulating SARS-CoV-2 Tropism Factor Expression by Inferring Cell-Type-Specific Transcriptional Regulatory Networks in Human Lungs. Viruses. 2022; 14(4):837. https://doi.org/10.3390/v14040837
Chicago/Turabian StyleTong, Haonan, Hao Chen, and Cranos M. Williams. 2022. "Identification of Transcription Factors Regulating SARS-CoV-2 Tropism Factor Expression by Inferring Cell-Type-Specific Transcriptional Regulatory Networks in Human Lungs" Viruses 14, no. 4: 837. https://doi.org/10.3390/v14040837