
Molecular Epidemiology and Evolution of Coxsackievirus A9

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Isolation and Nucleotide Sequencing
2.2. Datasets Construction of Worldwide and Chinese CVA9
2.3. Phylogenetic Analysis
2.4. Phylodynamic Analysis
2.5. Recombination Analysis
2.6. Assay for Temperature Sensitivity
2.7. Nucleotide Sequence Accession Numbers
3. Results
3.1. Virus Isolation
3.2. Dataset Description
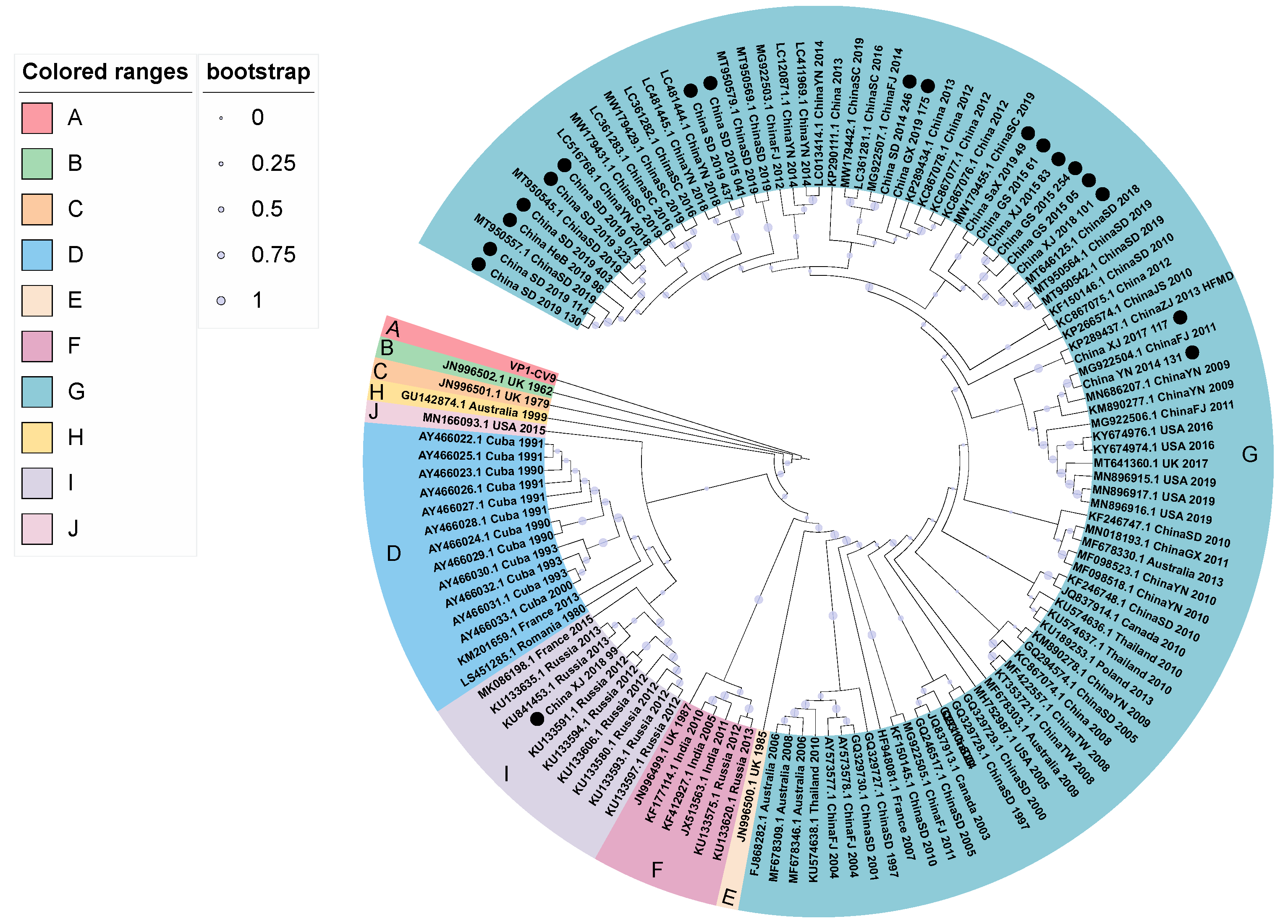
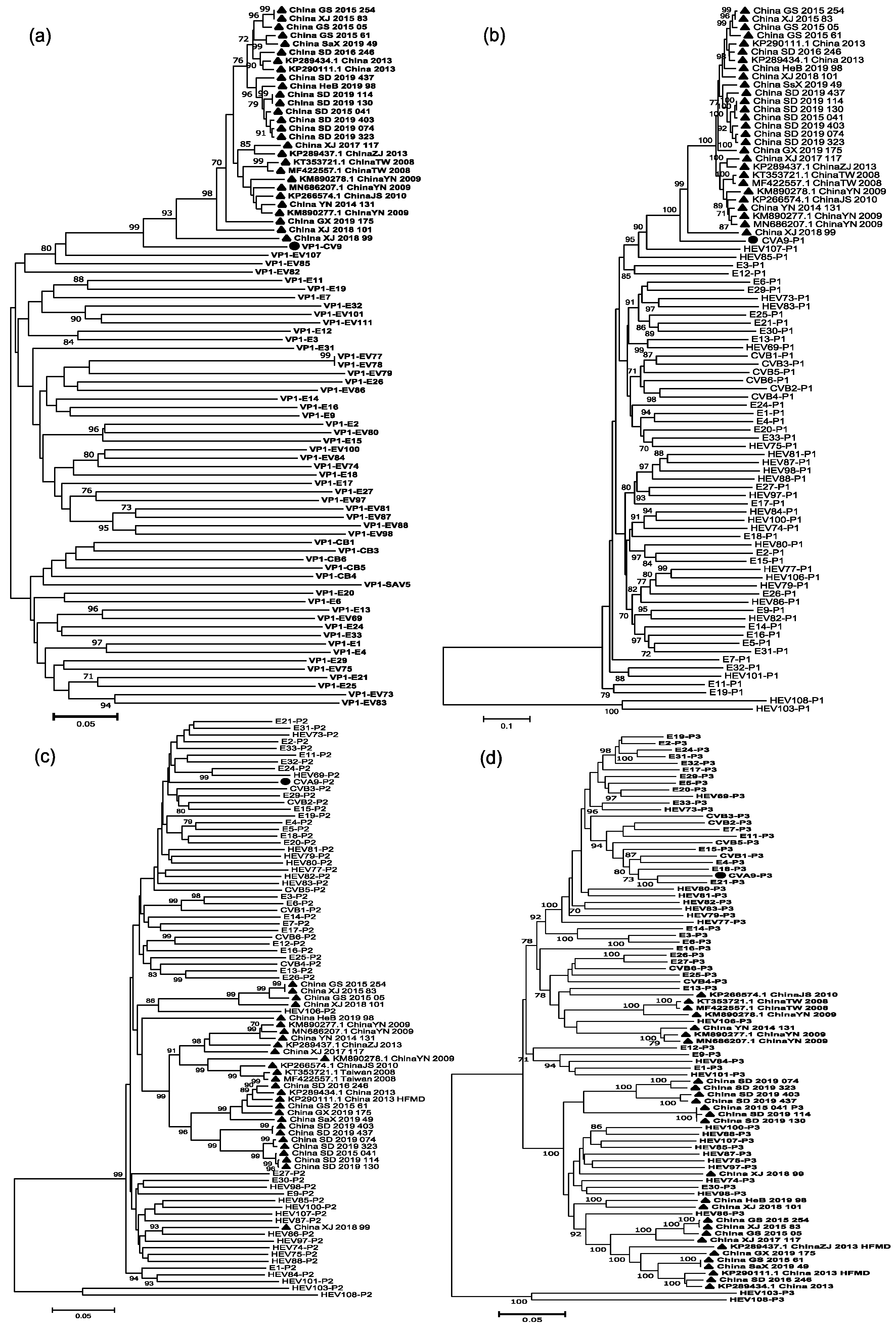
3.3. Phylogenetic Analysis
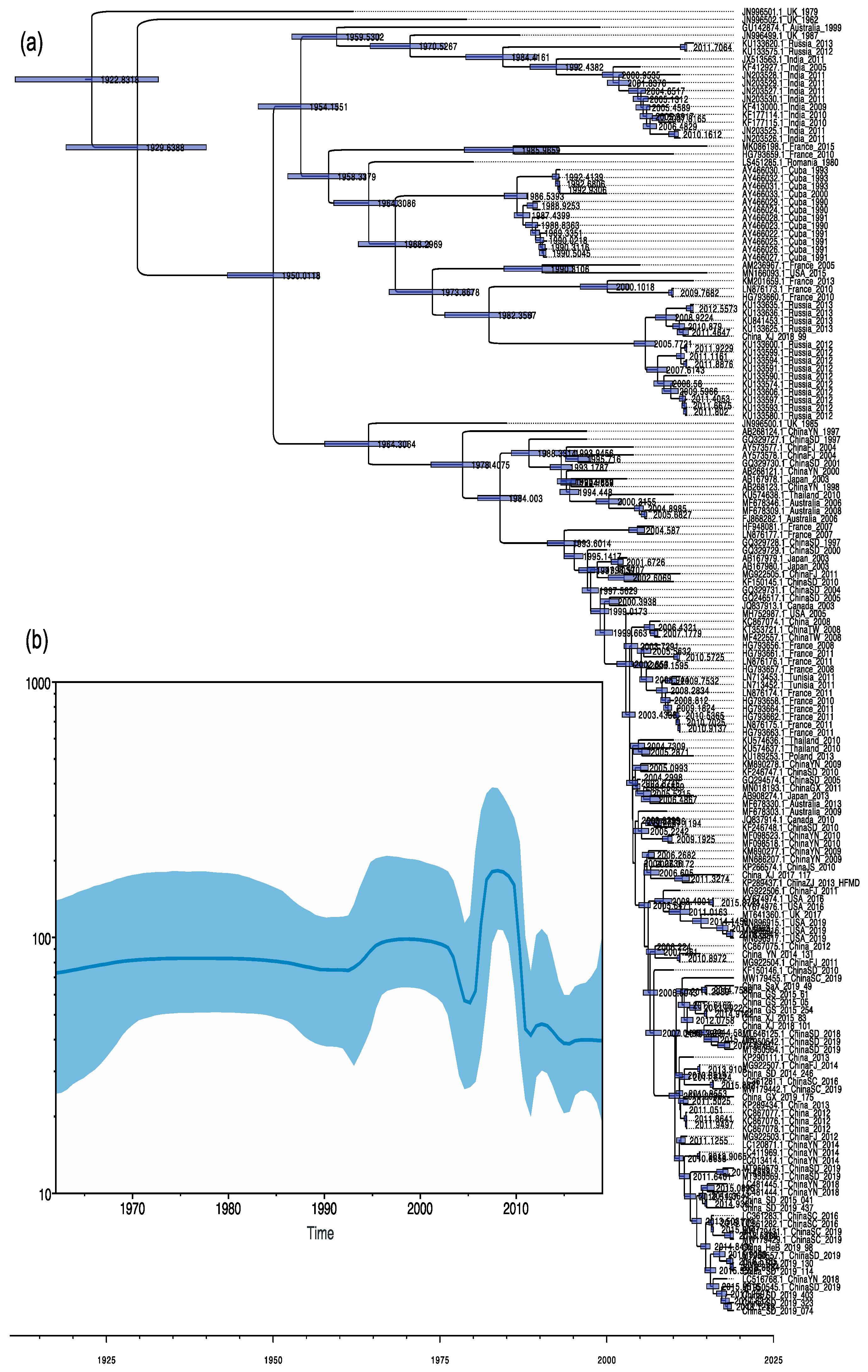
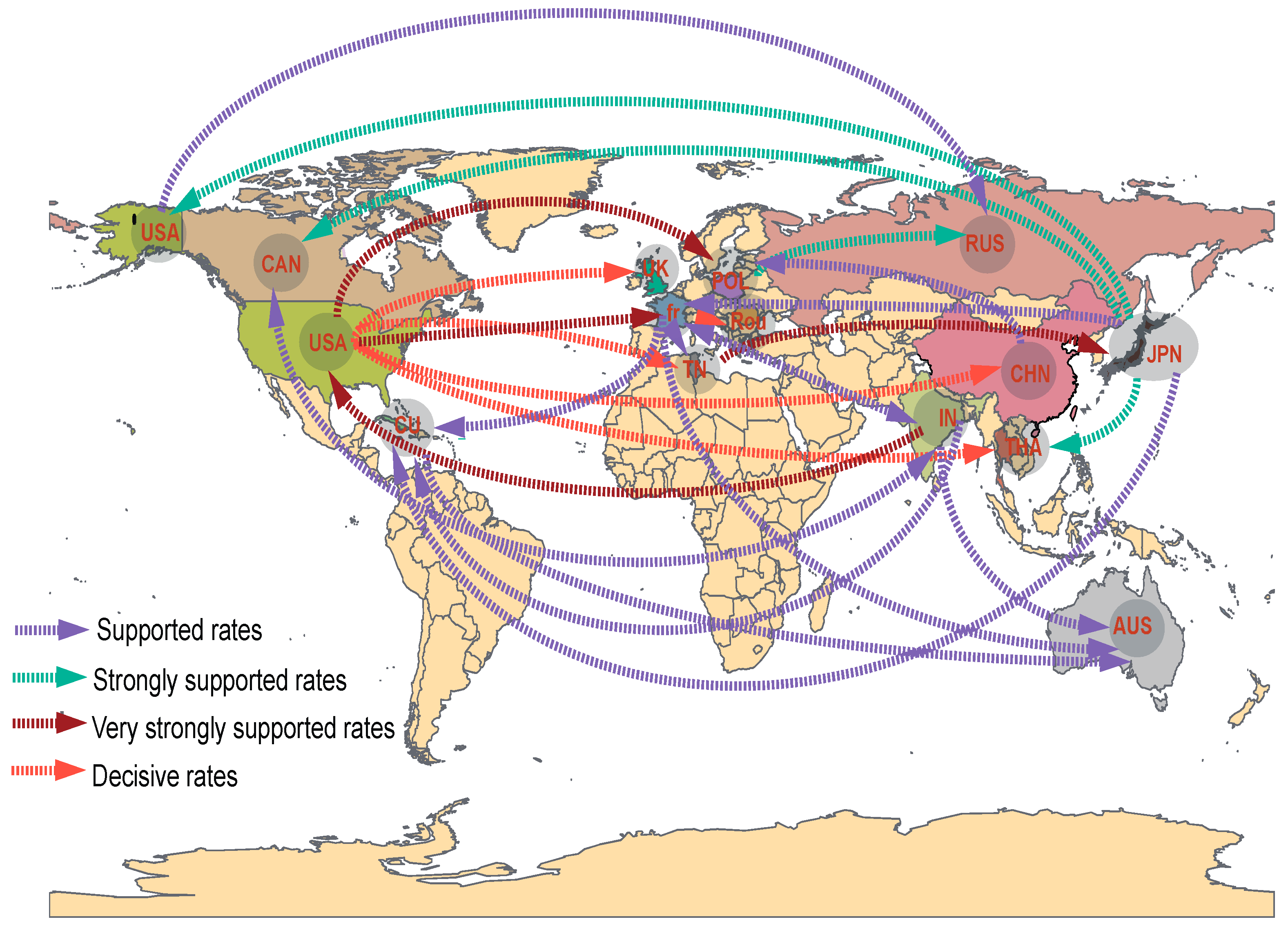
3.4. Phylodynamic Analysis
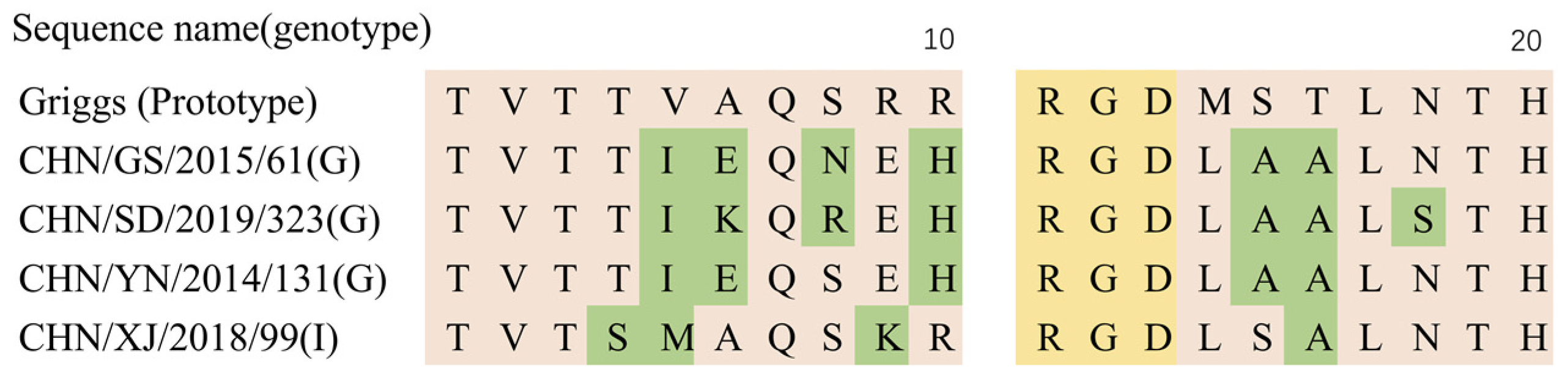
3.5. Analysis of Recombination Patterns of CVA9
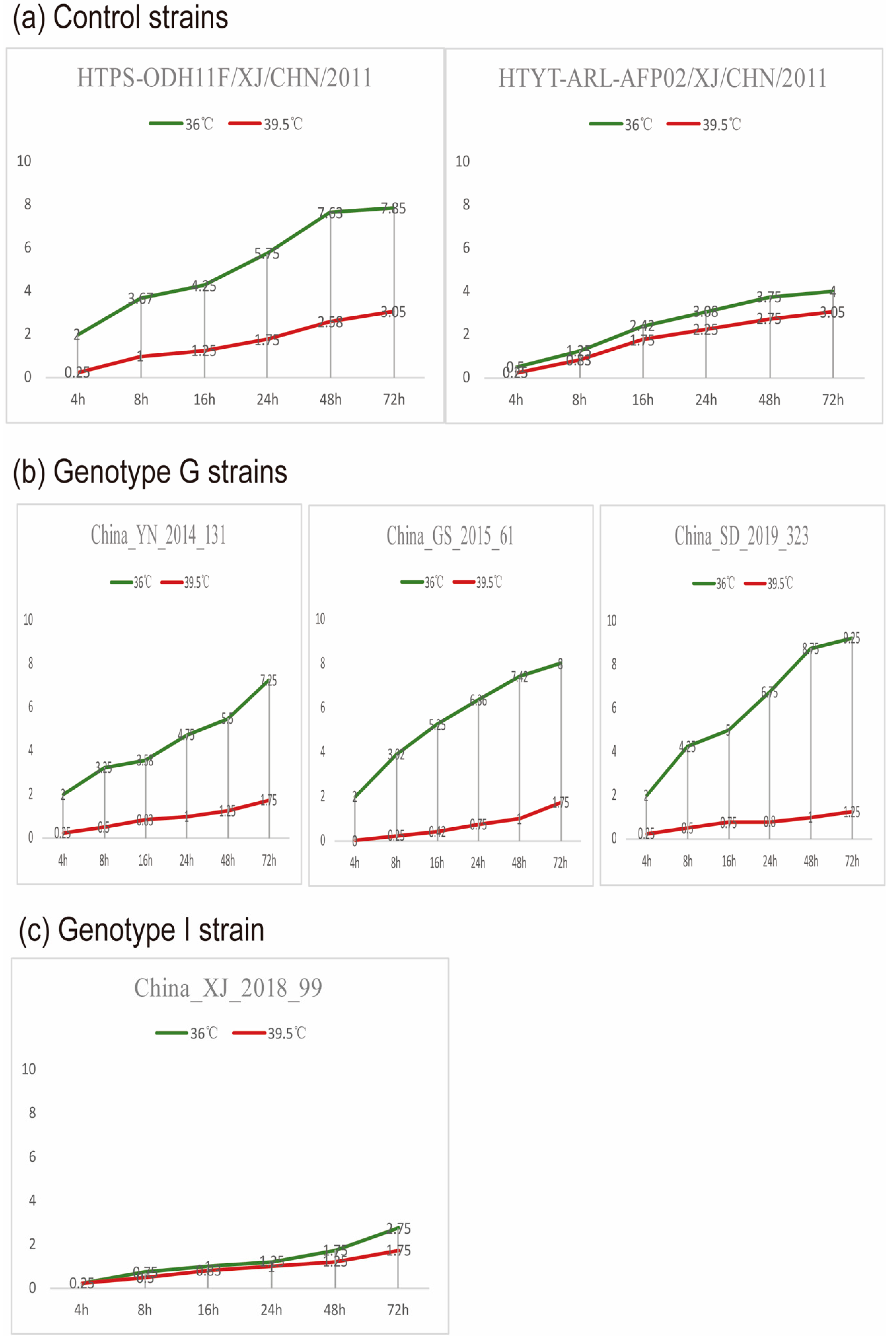
3.6. Temperature Sensitivity Properties of the CVA9
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simmonds, P.; Gorbalenya, A.E.; Harvala, H.; Hovi, T.; Knowles, N.J.; Lindberg, A.M.; Oberste, M.S.; Palmenberg, A.C.; Reuter, G.; Skern, T.; et al. Recommendations for the nomenclature of enteroviruses and rhinoviruses. Arch. Virol. 2020, 165, 793–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, A.M.Q.; Lefkowitz, E.J.; Mushegian, A.R.; Adams, M.J.; Dutilh, B.E.; Gorbalenya, A.E.; Harrach, B.; Harrison, R.L.; Junglen, S.; Knowles, N.J.; et al. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2018). Arch. Virol. 2018, 163, 2601–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.E.; Vogt, P.K. Cell entry by non-enveloped viruses. Curr. Top. Microbiol. Immunol. 2010, 343, v–vii. [Google Scholar] [PubMed]
- Merilahti, P.; Koskinen, S.; Heikkilä, O.; Karelehto, E.; Susi, P. Endocytosis of integrin-binding human picornaviruses. Adv. Virol. 2012, 2012, 547530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santti, J.; Harvala, H.; Kinnunen, L.; Hyypiä, T. Molecular epidemiology and evolution of coxsackievirus A9. J. Gen. Virol. 2000, 81 Pt 5, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Cui, A.; Yu, D.; Zhu, Z.; Meng, L.; Li, H.; Liu, J.; Liu, G.; Mao, N.; Xu, W. An outbreak of aseptic meningitis caused by coxsackievirus A9 in Gansu, the People’s Republic of China. Virol. J. 2010, 7, 72. [Google Scholar] [CrossRef] [Green Version]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Flemister, M.R.; Brown, B.A.; Pallansch, M.A. Typing of human enteroviruses by partial sequencing of VP1. J. Clin. Microbiol. 1999, 37, 1288–1293. [Google Scholar] [CrossRef] [Green Version]
- McWilliam Leitch, E.C.; Cabrerizo, M.; Cardosa, J.; Harvala, H.; Ivanova, O.E.; Koike, S.; Kroes, A.C.; Lukashev, A.; Perera, D.; Roivainen, M.; et al. The association of recombination events in the founding and emergence of subgenogroup evolutionary lineages of human enterovirus 71. J. Virol. 2012, 86, 2676–2685. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Y.; Chen, H.; Yan, D.M.; Zhang, Y.; An, J.J.; Ma, J.T.; Zhu, S.L.; Ma, X.M.; An, H.Q.; Xu, W.B. Genetic characterization of coxsackievirus A16 isolated in Ningxia Hui Municipality in 2008. Zhonghua Liu Xing Bing Xue Za Zhi 2010, 31, 904–908. [Google Scholar]
- He, Y.Q.; Chen, L.; Xu, W.B.; Yang, H.; Wang, H.Z.; Zong, W.P.; Xian, H.X.; Chen, H.L.; Yao, X.J.; Hu, Z.L.; et al. Emergence, circulation, and spatiotemporal phylogenetic analysis of coxsackievirus a6- and coxsackievirus a10-associated hand, foot, and mouth disease infections from 2008 to 2012 in Shenzhen, China. J. Clin. Microbiol. 2013, 51, 3560–3566. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kang, B.; Hwang, S.; Hong, J.; Chung, J.; Kim, S.; Jeong, Y.S.; Kim, K.; Cheon, D.S. Molecular characteristics of human coxsackievirus B1 infection in Korea, 2008–2009. J. Med. Virol. 2013, 85, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.A.; Oberste, M.S.; Alexander, J.P., Jr.; Kennett, M.L.; Pallansch, M.A. Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998. J. Virol. 1999, 73, 9969–9975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X. Chen.L.; Wang, H.; Jiang, M.; Zhu, H.; Xu, W.; Kong, X. Molecular Epidemiological Analysis of Coxsackievirus A9 in Children in Tibet Autonomous Region from 1999 to 2000. Chin. J. Exp. Clin. Virol. 2011, 1, 42–45. [Google Scholar]
- Xu, X. Chen.G.; Wang, Y.; An, R.; Shao, R.B. Molecular Evolutionary Features of VP1 of CVA9 from Hand, Foot and Mouth Disease in Yancheng City, 2016–2017. Jiangsu J. Prev. Med. 2019, 3, 269–271. [Google Scholar]
- Liu, J.; Zhu, Y.; Pan, Y.; Liu, Z.; Guo, C.; Ma, S. Complete genome sequence analysis of two human coxsackievirus A9 strains isolated in Yunnan, China, in 2009. Virus Genes 2015, 50, 358–364. [Google Scholar] [CrossRef]
- Drake, J.W. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 1993, 90, 4171–4175. [Google Scholar] [CrossRef] [Green Version]
- Cardosa, M.J.; Perera, D.; Brown, B.A.; Cheon, D.; Chan, H.M.; Chan, K.P.; Cho, H.; McMinn, P. Molecular epidemiology of human enterovirus 71 strains and recent outbreaks in the Asia-Pacific region: Comparative analysis of the VP1 and VP4 genes. Emerg. Infect. Dis. 2003, 9, 461–468. [Google Scholar] [CrossRef]
- Woodman, A.; Arnold, J.J.; Cameron, C.E.; Evans, D.J. Biochemical and genetic analysis of the role of the viral polymerase in enterovirus recombination. Nucleic Acids Res. 2016, 44, 6883–6895. [Google Scholar] [CrossRef]
- Pybus, O.G.; Suchard, M.A.; Lemey, P.; Bernardin, F.J.; Rambaut, A.; Crawford, F.W.; Gray, R.R.; Arinaminpathy, N.; Stramer, S.L.; Busch, M.P.; et al. Unifying the spatial epidemiology and molecular evolution of emerging epidemics. Proc. Natl. Acad. Sci. USA 2012, 109, 15066–15071. [Google Scholar] [CrossRef] [Green Version]
- Allicock, O.M.; Lemey, P.; Tatem, A.J.; Pybus, O.G.; Bennett, S.N.; Mueller, B.A.; Suchard, M.A.; Foster, J.E.; Rambaut, A.; Carrington, C.V. Phylogeography and population dynamics of dengue viruses in the Americas. Mol. Biol. Evol. 2012, 29, 1533–1543. [Google Scholar] [CrossRef] [Green Version]
- Khetsuriani, N.; Lamonte-Fowlkes, A.; Oberst, S.; Pallansch, M.A. Enterovirus surveillance—United States, 1970–2005. MMWR Surveill. Summ. 2006, 55, 1–20. [Google Scholar] [PubMed]
- Huang, Y.C.; Chu, Y.H.; Yen, T.Y.; Huang, W.C.; Huang, L.M.; Cheng, A.L.; Wang, H.Y.; Chang, L.Y. Clinical features and phylogenetic analysis of Coxsackievirus A9 in Northern Taiwan in 2011. BMC Infect. Dis. 2013, 13, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pabbaraju, K.; Wong, S.; Chan, E.N.; Tellier, R. Genetic characterization of a Coxsackie A9 virus associated with aseptic meningitis in Alberta, Canada in 2010. Virol. J. 2013, 10, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Z.; Wang, H.; Li, Y.; Liu, G.; Xu, A.; Lin, X.; Song, L.; Ji, F.; Wang, S.; Cui, N.; et al. Molecular epidemiology of human enterovirus associated with aseptic meningitis in Shandong Province, China, 2006–2012. PLoS ONE 2014, 9, e89766. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wang, H.; Tao, Z.; Xu, A.; Lin, X.; Zhou, N.; Wang, P.; Wang, Q. Multiple transmission chains of coxsackievirus A4 co-circulating in China and neighboring countries in recent years: Phylogenetic and spatiotemporal analyses based on virological surveillance. Mol. Phylogenet. Evol. 2018, 118, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Zhang, Y. Isolation and Characterization of Vaccine-Derived Polioviruses, Relevance for the Global Polio Eradication Initiative. Methods Mol. Biol. 2016, 1387, 213–226. [Google Scholar]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Khoosal, A.; Muhire, B. Detecting and Analyzing Genetic Recombination Using RDP4. Methods Mol. Biol. 2017, 1525, 433–460. [Google Scholar]
- Song, Y.; Zhang, Y.; Fan, Q.; Cui, H.; Yan, D.; Zhu, S.; Tang, H.; Sun, Q.; Wang, D.; Xu, W. Phylogenetic Characterizations of Highly Mutated EV-B106 Recombinants Showing Extensive Genetic Exchanges with Other EV-B in Xinjiang, China. Sci. Rep. 2017, 7, 43080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Zhang, Y.; Zhu, S.; Tian, H.; Huang, G.; Cui, H.; Li, X.; Yan, D.; Zhu, Z.; Li, J.; et al. Transmission of human enterovirus 85 recombinants containing new unknown serotype HEV-B donor sequences in Xinjiang Uighur autonomous region, China. PLoS ONE 2013, 8, e55480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Pallansch, M.A. Molecular evolution of the human enteroviruses: Correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 1999, 73, 1941–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Othman, I.; Mirand, A.; Slama, I.; Mastouri, M.; Peigue-Lafeuille, H.; Aouni, M.; Bailly, J.L. Enterovirus Migration Patterns between France and Tunisia. PLoS ONE 2015, 10, e0145674. [Google Scholar] [CrossRef] [PubMed]
- Sanders, B.P.; de Los Rios Oakes, I.; van Hoek, V.; Bockstal, V.; Kamphuis, T.; Uil, T.G.; Song, Y.; Cooper, G.; Crawt, L.E.; Martín, J.; et al. Cold-Adapted Viral Attenuation (CAVA): Highly Temperature Sensitive Polioviruses as Novel Vaccine Strains for a Next Generation Inactivated Poliovirus Vaccine. PLoS Pathog. 2016, 12, e1005483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hietanen, E.; Susi, P. Recombination Events and Conserved Nature of Receptor Binding Motifs in Coxsackievirus A9 Isolates. Viruses 2020, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.K.; Simmonds, P.; Harvala, H. The importance of enterovirus surveillance in a post-polio world. Lancet Infect. Dis. 2021, 22, e35–e40. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pattern | Region | Name of Strains or GenBank Accession | Representative Strain |
|---|---|---|---|
| No recombination | N/A | MF42255, KT352721, GX19–175 | N/A |
| RF1 | P2, P3 | KP289434, KP290111, KP289437, SD/2016/246, XJ/2017/117, SaX/2019/49 | XJ/2017/117 |
| RF2 | P2 | HeB/2019/98, XJ/2018/101 | HeB/2019/98 |
| RF3 | P2 | YN/2014/131, KP266574, MN686207, KM890277 | YN/2014/131 |
| RF4 | 5′, P2 | SD/2019/074, SD/2019/323, SD/2019/403, SD/2019/437, SD/2015/041 | SD/2019/403 |
| RF5 | 5′, P2, P3 | SD/2019/114, SD/2019/130 | SD/2019/130 |
| RF6 | P2, P3 | XJ/2015/83, GS/2015/05, GS/2015/61, GS/2015/254 | GS/2015/61 |
| RF7 | P2, P3 | XJ/2018/99 | XJ/2018/99 |
| RF8 | P3 | KM890278 | KM890278 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, H.; Wang, J.; Chen, J.; Huang, R.; Zhang, Y.; Xiao, J.; Song, Y.; Ji, T.; Yang, Q.; Zhu, S.; et al. Molecular Epidemiology and Evolution of Coxsackievirus A9. Viruses 2022, 14, 822. https://doi.org/10.3390/v14040822
Zhao H, Wang J, Chen J, Huang R, Zhang Y, Xiao J, Song Y, Ji T, Yang Q, Zhu S, et al. Molecular Epidemiology and Evolution of Coxsackievirus A9. Viruses. 2022; 14(4):822. https://doi.org/10.3390/v14040822
Chicago/Turabian StyleZhao, Hehe, Jianxing Wang, Jianhua Chen, Ruifang Huang, Yong Zhang, Jinbo Xiao, Yang Song, Tianjiao Ji, Qian Yang, Shuangli Zhu, and et al. 2022. "Molecular Epidemiology and Evolution of Coxsackievirus A9" Viruses 14, no. 4: 822. https://doi.org/10.3390/v14040822