
TP53 Gene Therapy as a Potential Treatment for Patients with COVID-19

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of p53 in Immunity
3. Evidence for p53 Involvement in Broadly Opposing Infections by Viruses
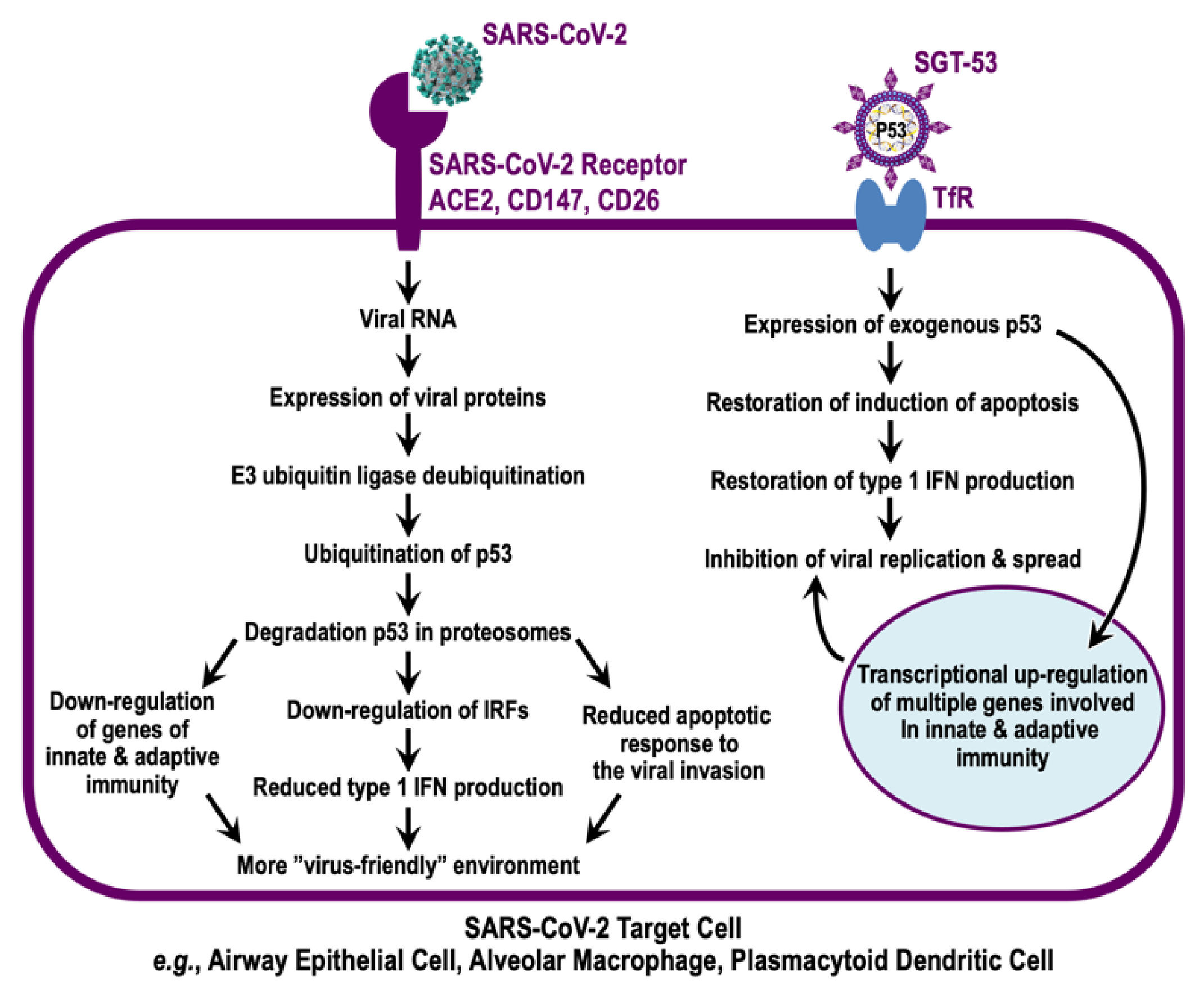
4. Evidence for p53 Involvement in Combatting Infections by Coronaviruses
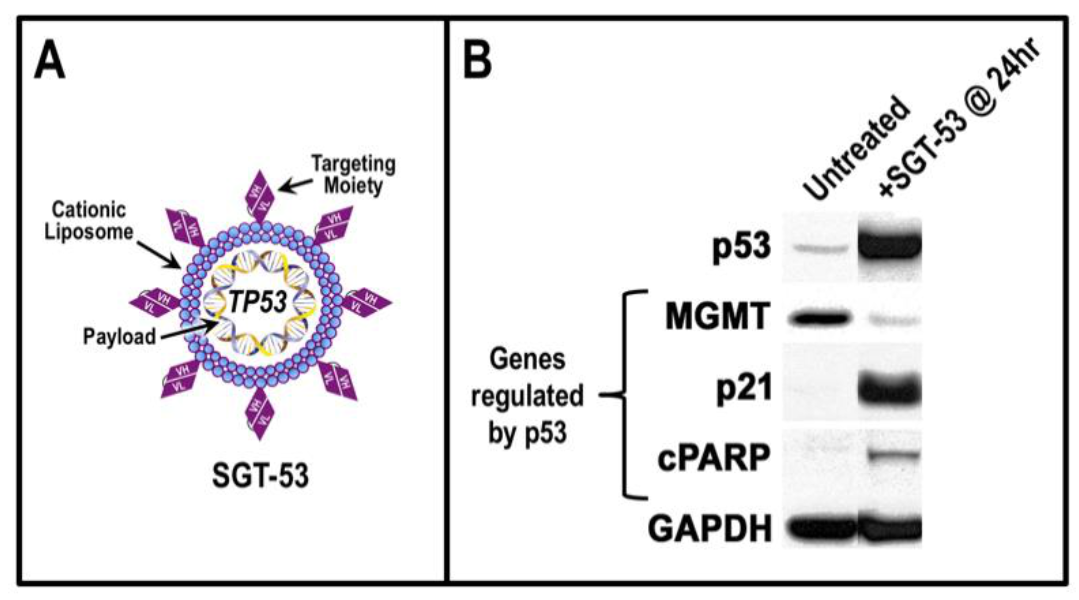
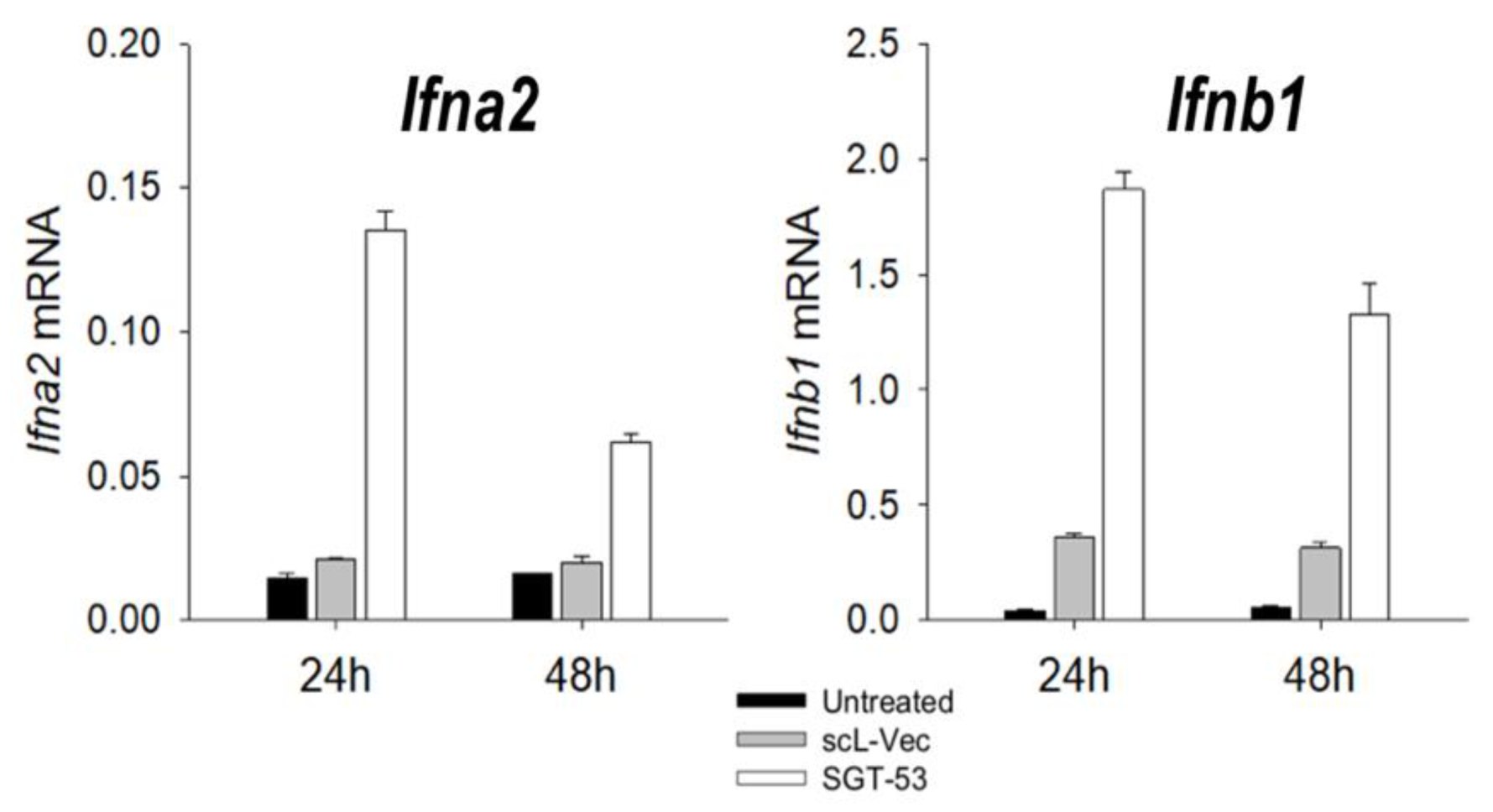
5. TP53 Gene Therapy as an Antiviral Strategy
6. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, M.Y.; Zhao, R.; Gao, L.J.; Gao, X.F.; Wang, D.P.; Cao, J.M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell. Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ho, W.; Huang, Y.; Jin, D.Y.; Li, S.; Liu, S.L.; Liu, X.; Qiu, J.; Sang, Y.; Wang, Q.; et al. SARS-CoV-2 is an appropriate name for the new coronavirus. Lancet 2020, 395, 949–950. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Rivas, C.; Aaronson, S.A.; Munoz-Fontela, C. Dual Role of p53 in Innate Antiviral Immunity. Viruses 2010, 2, 298–313. [Google Scholar] [CrossRef] [Green Version]
- Lazo, P.A.; Santos, C.R. Interference with p53 functions in human viral infections, a target for novel antiviral strategies? Rev. Med. Virol. 2011, 21, 285–300. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Tsurumi, T. Genome guardian p53 and viral infections. Rev. Med. Virol. 2013, 23, 213–220. [Google Scholar] [CrossRef]
- Aloni-Grinstein, R.; Charni-Natan, M.; Solomon, H.; Rotter, V. p53 and the Viral Connection: Back into the Future (double dagger). Cancers 2018, 10, 178. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. P53 and The Immune Response: 40 Years of Exploration-A Plan for the Future. Int. J. Mol. Sci. 2020, 21, 541. [Google Scholar] [CrossRef] [Green Version]
- Cardozo, C.M.; Hainaut, P. Viral strategies for circumventing p53: The case of severe acute respiratory syndrome coronavirus. Curr. Opin. Oncol. 2021, 33, 149–158. [Google Scholar] [CrossRef]
- Yuan, L.; Chen, Z.; Song, S.; Wang, S.; Tian, C.; Xing, G.; Chen, X.; Xiao, Z.X.; He, F.; Zhang, L. p53 degradation by a coronavirus papain-like protease suppresses type I interferon signaling. J. Biol. Chem. 2015, 290, 3172–3182. [Google Scholar] [CrossRef] [Green Version]
- Ma-Lauer, Y.; Carbajo-Lozoya, J.; Hein, M.Y.; Muller, M.A.; Deng, W.; Lei, J.; Meyer, B.; Kusov, Y.; von Brunn, B.; Bairad, D.R.; et al. p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc. Natl. Acad. Sci. USA 2016, 113, E5192–E5201. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb. Perspect. Med. 2016, 6, a026179. [Google Scholar] [CrossRef] [Green Version]
- Miciak, J.; Bunz, F. Long story short: p53 mediates innate immunity. Biochim. Biophys. Acta 2016, 1865, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Blagih, J.; Buck, M.D.; Vousden, K.H. p53, cancer and the immune response. J. Cell Sci. 2020, 133, jcs237453. [Google Scholar] [CrossRef] [Green Version]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Munoz-Fontela, C.; Garcia, M.A.; Garcia-Cao, I.; Collado, M.; Arroyo, J.; Esteban, M.; Serrano, M.; Rivas, C. Resistance to viral infection of super p53 mice. Oncogene 2005, 24, 3059–3062. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Fontela, C.; Pazos, M.; Delgado, I.; Murk, W.; Mungamuri, S.K.; Lee, S.W.; Garcia-Sastre, A.; Moran, T.M.; Aaronson, S.A. p53 serves as a host antiviral factor that enhances innate and adaptive immune responses to influenza A virus. J. Immunol. 2011, 187, 6428–6436. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Bargonetti, J.; Reynisdottir, I.; Friedman, P.N.; Prives, C. Site-specific binding of wild-type p53 to cellular DNA is inhibited by SV40 T antigen and mutant p53. Genes Dev. 1992, 6, 1886–1898. [Google Scholar] [CrossRef] [Green Version]
- Yew, P.R.; Liu, X.; Berk, A.J. Adenovirus E1B oncoprotein tethers a transcriptional repression domain to p53. Genes Dev. 1994, 8, 190–202. [Google Scholar] [CrossRef] [Green Version]
- Terrier, O.; Diederichs, A.; Dubois, J.; Cartet, G.; Lina, B.; Bourdon, J.C.; Rosa-Calatrava, M. Influenza NS1 interacts with p53 and alters its binding to p53-responsive genes, in a promoter-dependent manner. FEBS Lett. 2013, 587, 2965–2971. [Google Scholar] [CrossRef] [Green Version]
- Crook, T.; Tidy, J.A.; Vousden, K.H. Degradation of p53 can be targeted by HPV E6 sequences distinct from those required for p53 binding and trans-activation. Cell 1991, 67, 547–556. [Google Scholar] [CrossRef]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Teodoro, J.G.; Branton, P.E. Regulation of apoptosis by viral gene products. J. Virol. 1997, 71, 1739–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedelcu, A.M.; Driscoll, W.W.; Durand, P.M.; Herron, M.D.; Rashidi, A. On the paradigm of altruistic suicide in the unicellular world. Evolution 2011, 65, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Forrester, K.; Yeh, H.; Feitelson, M.A.; Gu, J.R.; Harris, C.C. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc. Natl. Acad. Sci. USA 1994, 91, 2230–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Schwedes, J.F.; Parks, D.; Mann, K.; Tegtmeyer, P. Interaction of p53 with its consensus DNA-binding site. Mol. Cell. Biol. 1995, 15, 2157–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Turpin, E.; Luke, K.; Jones, J.; Tumpey, T.; Konan, K.; Schultz-Cherry, S. Influenza virus infection increases p53 activity: Role of p53 in cell death and viral replication. J. Virol. 2005, 79, 8802–8811. [Google Scholar] [CrossRef] [Green Version]
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Honda, K.; et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003, 424, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Fontela, C.; Macip, S.; Martinez-Sobrido, L.; Brown, L.; Ashour, J.; Garcia-Sastre, A.; Lee, S.W.; Aaronson, S.A. Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med. 2008, 205, 1929–1938. [Google Scholar] [CrossRef] [Green Version]
- Hemmat, N.; Asadzadeh, Z.; Ahangar, N.K.; Alemohammad, H.; Najafzadeh, B.; Derakhshani, A.; Baghbanzadeh, A.; Baghi, H.B.; Javadrashid, D.; Najafi, S.; et al. The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV. Arch. Virol. 2021, 166, 675–696. [Google Scholar] [CrossRef]
- Sa Ribero, M.; Jouvenet, N.; Dreux, M.; Nisole, S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog. 2020, 16, e1008737. [Google Scholar] [CrossRef]
- Sallard, E.; Lescure, F.X.; Yazdanpanah, Y.; Mentre, F.; Peiffer-Smadja, N. Type 1 interferons as a potential treatment against COVID-19. Antivir. Res. 2020, 178, 104791. [Google Scholar] [CrossRef]
- Lu, H. Drug treatment options for the 2019-new coronavirus (2019-nCoV). Biosci. Trends 2020, 14, 69–71. [Google Scholar] [CrossRef] [Green Version]
- Paces, J.; Strizova, Z.; Smrz, D.; Cerny, J. COVID-19 and the immune system. Physiol. Res. 2020, 69, 379–388. [Google Scholar] [CrossRef]
- Smits, S.L.; de Lang, A.; van den Brand, J.M.; Leijten, L.M.; van, I.W.F.; Eijkemans, M.J.; van Amerongen, G.; Kuiken, T.; Andeweg, A.C.; Osterhaus, A.D.; et al. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. 2010, 6, e1000756. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 533–535. [Google Scholar] [CrossRef] [Green Version]
- Pribul, P.K.; Harker, J.; Wang, B.; Wang, H.; Tregoning, J.S.; Schwarze, J.; Openshaw, P.J. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J. Virol. 2008, 82, 4441–4448. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Christman, J.W. Editorial: Alveolar Macrophages in Lung Inflammation and Resolution. Front. Immunol. 2019, 10, 2275. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, Y.; Takeuchi, O.; Kato, H.; Kumar, H.; Matsui, K.; Morii, E.; Aozasa, K.; Kawai, T.; Akira, S. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity 2007, 27, 240–252. [Google Scholar] [CrossRef] [Green Version]
- Divangahi, M.; King, I.L.; Pernet, E. Alveolar macrophages and type I IFN in airway homeostasis and immunity. Trends Immunol. 2015, 36, 307–314. [Google Scholar] [CrossRef]
- Goritzka, M.; Makris, S.; Kausar, F.; Durant, L.R.; Pereira, C.; Kumagai, Y.; Culley, F.J.; Mack, M.; Akira, S.; Johansson, C. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J. Exp. Med. 2015, 212, 699–714. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Radzikowska, U.; Ding, M.; Tan, G.; Zhakparov, D.; Peng, Y.; Wawrzyniak, P.; Wang, M.; Li, S.; Morita, H.; Altunbulakli, C.; et al. Distribution of ACE2, CD147, CD26, and other SARS-CoV-2 associated molecules in tissues and immune cells in health and in asthma, COPD, obesity, hypertension, and COVID-19 risk factors. Allergy 2020, 75, 2829–2845. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Chu, H.; Chan, J.F.; Wang, Y.; Yuen, T.T.; Chai, Y.; Hou, Y.; Shuai, H.; Yang, D.; Hu, B.; Huang, X.; et al. Comparative Replication and Immune Activation Profiles of SARS-CoV-2 and SARS-CoV in Human Lungs: An Ex Vivo Study With Implications for the Pathogenesis of COVID-19. Clin. Infect. Dis. 2020, 71, 1400–1409. [Google Scholar] [CrossRef] [Green Version]
- Dalskov, L.; Mohlenberg, M.; Thyrsted, J.; Blay-Cadanet, J.; Poulsen, E.T.; Folkersen, B.H.; Skaarup, S.H.; Olagnier, D.; Reinert, L.; Enghild, J.J.; et al. SARS-CoV-2 evades immune detection in alveolar macrophages. EMBO Rep. 2020, 21, e51252. [Google Scholar] [CrossRef]
- de Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Webster, B.; Assil, S.; Dreux, M. Cell-Cell Sensing of Viral Infection by Plasmacytoid Dendritic Cells. J. Virol. 2016, 90, 10050–10053. [Google Scholar] [CrossRef] [Green Version]
- Reizis, B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019, 50, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Cervantes-Barragan, L.; Zust, R.; Weber, F.; Spiegel, M.; Lang, K.S.; Akira, S.; Thiel, V.; Ludewig, B. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood 2007, 109, 1131–1137. [Google Scholar] [CrossRef] [Green Version]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Grunewald, M.; Perlman, S. Coronaviruses: An Updated Overview of Their Replication and Pathogenesis. Methods Mol. Biol. 2020, 2203, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Clementz, M.A.; Chen, Z.; Banach, B.S.; Wang, Y.; Sun, L.; Ratia, K.; Baez-Santos, Y.M.; Wang, J.; Takayama, J.; Ghosh, A.K.; et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010, 84, 4619–4629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Hu, W.; Yen, Y.T.; Singh, S.; Kao, C.L.; Wu-Hsieh, B.A. SARS-CoV regulates immune function-related gene expression in human monocytic cells. Viral Immunol. 2012, 25, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Cummins, J.M.; Vogelstein, B. HAUSP is required for p53 destabilization. Cell Cycle 2004, 3, 689–692. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Zauli, G.; Tisato, V.; Secchiero, P. Rationale for Considering Oral Idasanutlin as a Therapeutic Option for COVID-19 Patients. Front. Pharmacol. 2020, 11, 1156. [Google Scholar] [CrossRef]
- Lee, J.H.; Chung, H.C.; Nguyen, V.G.; Moon, H.J.; Kim, H.K.; Park, S.J.; Lee, C.H.; Lee, G.E.; Park, B.K. Detection and Phylogenetic Analysis of Porcine Deltacoronavirus in Korean Swine Farms, 2015. Transbound. Emerg. Dis. 2016, 63, 248–252. [Google Scholar] [CrossRef]
- Sun, P.; Wu, H.; Huang, J.; Xu, Y.; Yang, F.; Zhang, Q.; Xu, X. Porcine epidemic diarrhea virus through p53-dependent pathway causes cell cycle arrest in the G0/G1 phase. Virus Res. 2018, 253, 1–11. [Google Scholar] [CrossRef]
- Xing, Y.; Chen, J.; Tu, J.; Zhang, B.; Chen, X.; Shi, H.; Baker, S.C.; Feng, L.; Chen, Z. The papain-like protease of porcine epidemic diarrhea virus negatively regulates type I interferon pathway by acting as a viral deubiquitinase. J. Gen. Virol. 2013, 94, 1554–1567. [Google Scholar] [CrossRef]
- Hao, Z.; Fu, F.; Cao, L.; Guo, L.; Liu, J.; Xue, M.; Feng, L. Tumor suppressor p53 inhibits porcine epidemic diarrhea virus infection via interferon-mediated antiviral immunity. Mol. Immunol. 2019, 108, 68–74. [Google Scholar] [CrossRef]
- Garcia-Cao, I.; Garcia-Cao, M.; Martin-Caballero, J.; Criado, L.M.; Klatt, P.; Flores, J.M.; Weill, J.C.; Blasco, M.A.; Serrano, M. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002, 21, 6225–6235. [Google Scholar] [CrossRef] [Green Version]
- Daniels, T.R.; Bernabeu, E.; Rodriguez, J.A.; Patel, S.; Kozman, M.; Chiappetta, D.A.; Holler, E.; Ljubimova, J.Y.; Helguera, G.; Penichet, M.L. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim. Biophys. Acta 2012, 1820, 291–317. [Google Scholar] [CrossRef] [Green Version]
- Paterson, J.; Webster, C.I. Exploiting transferrin receptor for delivering drugs across the blood-brain barrier. Drug Discov. Today Technol. 2016, 20, 49–52. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Delivery of Biologics Across the Blood-Brain Barrier with Molecular Trojan Horse Technology. BioDrugs 2017, 31, 503–519. [Google Scholar] [CrossRef]
- Johnsen, K.B.; Burkhart, A.; Thomsen, L.B.; Andresen, T.L.; Moos, T. Targeting the transferrin receptor for brain drug delivery. Prog. Neurobiol. 2019, 181, 101665. [Google Scholar] [CrossRef]
- Kim, S.S.; Harford, J.B.; Pirollo, K.F.; Chang, E.H. Effective treatment of glioblastoma requires crossing the blood-brain barrier and targeting tumors including cancer stem cells: The promise of nanomedicine. Biochem. Biophys. Res. Commun. 2015, 468, 485–489. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.S.; Rait, A.; Kim, E.; Pirollo, K.F.; Nishida, M.; Farkas, N.; Dagata, J.A.; Chang, E.H. A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival. ACS Nano 2014, 8, 5494–5514. [Google Scholar] [CrossRef]
- Xu, L.; Pirollo, K.F.; Chang, E.H. Tumor-targeted p53-gene therapy enhances the efficacy of conventional chemo/radiotherapy. J. Control. Release 2001, 74, 115–128. [Google Scholar] [CrossRef]
- Moore, E.C.; Sun, L.; Clavijo, P.E.; Friedman, J.; Harford, J.B.; Saleh, A.D.; Van Waes, C.; Chang, E.H.; Allen, C.T. Nanocomplex-based TP53 gene therapy promotes anti-tumor immunity through TP53- and STING-dependent mechanisms. Oncoimmunology 2018, 7, e1404216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.S.; Harford, J.B.; Moghe, M.; Rait, A.; Chang, E.H. Combination with SGT-53 overcomes tumor resistance to a checkpoint inhibitor. Oncoimmunology 2018, 7, e1484982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.S.; Harford, J.B.; Moghe, M.; Slaughter, T.; Doherty, C.; Chang, E.H. A tumor-targeting nanomedicine carrying the p53 gene crosses the blood-brain barrier and enhances anti-PD-1 immunotherapy in mouse models of glioblastoma. Int. J. Cancer 2019, 145, 2535–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senzer, N.; Nemunaitis, J.; Nemunaitis, D.; Bedell, C.; Edelman, G.; Barve, M.; Nunan, R.; Pirollo, K.F.; Rait, A.; Chang, E.H. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol. Ther. 2013, 21, 1096–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirollo, K.F.; Nemunaitis, J.; Leung, P.K.; Nunan, R.; Adams, J.; Chang, E.H. Safety and Efficacy in Advanced Solid Tumors of a Targeted Nanocomplex Carrying the p53 Gene Used in Combination with Docetaxel: A Phase 1b Study. Mol. Ther. 2016, 24, 1697–1706. [Google Scholar] [CrossRef] [Green Version]
- van der Meel, R.; Vehmeijer, L.J.; Kok, R.J.; Storm, G.; van Gaal, E.V. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: Current status. Adv. Drug Deliv. Rev. 2013, 65, 1284–1298. [Google Scholar] [CrossRef]
- Biffi, S.; Voltan, R.; Bortot, B.; Zauli, G.; Secchiero, P. Actively targeted nanocarriers for drug delivery to cancer cells. Expert Opin. Drug Deliv. 2019, 16, 481–496. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Vareille, M.; Kieninger, E.; Edwards, M.R.; Regamey, N. The airway epithelium: Soldier in the fight against respiratory viruses. Clin. Microbiol. Rev. 2011, 24, 210–229. [Google Scholar] [CrossRef] [Green Version]
- Carcaterra, M.; Caruso, C. Alveolar epithelial cell type II as main target of SARS-CoV-2 virus and COVID-19 development via NF-Kb pathway deregulation: A physio-pathological theory. Med. Hypotheses 2021, 146, 110412. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, C.F.; Kuhn, J.H.; Nguyen, D.; Li, W.; Nagel, J.; Schmidt, P.J.; Nunberg, J.H.; Andrews, N.C.; et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar] [CrossRef]
- Flanagan, M.L.; Oldenburg, J.; Reignier, T.; Holt, N.; Hamilton, G.A.; Martin, V.K.; Cannon, P.M. New world clade B arenaviruses can use transferrin receptor 1 (TfR1)-dependent and -independent entry pathways, and glycoproteins from human pathogenic strains are associated with the use of TfR1. J. Virol. 2008, 82, 938–948. [Google Scholar] [CrossRef] [Green Version]
- Andreesen, R.; Osterholz, J.; Bodemann, H.; Bross, K.J.; Costabel, U.; Lohr, G.W. Expression of transferrin receptors and intracellular ferritin during terminal differentiation of human monocytes. Blut 1984, 49, 195–202. [Google Scholar] [CrossRef]
- Hirata, T.; Bitterman, P.B.; Mornex, J.F.; Crystal, R.G. Expression of the transferrin receptor gene during the process of mononuclear phagocyte maturation. J. Immunol. 1986, 136, 1339–1345. [Google Scholar]
- Lippitsch, A.; Chukovetskyi, Y.; Baal, N.; Bein, G.; Hackstein, H. Unique high and homogenous surface expression of the transferrin receptor CD71 on murine plasmacytoid dendritic cells in different tissues. Cell. Immunol. 2017, 316, 41–52. [Google Scholar] [CrossRef]
- Coronavirus Resource Center. Available online: https://coronavirus.jhu.edu (accessed on 13 March 2022).
- Kim, S.S.; Rait, A.; Rubab, F.; Rao, A.K.; Kiritsy, M.C.; Pirollo, K.F.; Wang, S.; Weiner, L.M.; Chang, E.H. The clinical potential of targeted nanomedicine: Delivering to cancer stem-like cells. Mol. Ther. 2014, 22, 278–291. [Google Scholar] [CrossRef] [Green Version]
- Abegglen, L.M.; Caulin, A.F.; Chan, A.; Lee, K.; Robinson, R.; Campbell, M.S.; Kiso, W.K.; Schmitt, D.L.; Waddell, P.J.; Bhaskara, S.; et al. Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA 2015, 314, 1850–1860. [Google Scholar] [CrossRef]
- Salama, C.; Han, J.; Yau, L.; Reiss, W.G.; Kramer, B.; Neidhart, J.D.; Criner, G.J.; Kaplan-Lewis, E.; Baden, R.; Pandit, L.; et al. Tocilizumab in Patients Hospitalized with COVID-19 Pneumonia. N. Engl. J. Med. 2021, 384, 20–30. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harford, J.B.; Kim, S.S.; Pirollo, K.F.; Chang, E.H. TP53 Gene Therapy as a Potential Treatment for Patients with COVID-19. Viruses 2022, 14, 739. https://doi.org/10.3390/v14040739
Harford JB, Kim SS, Pirollo KF, Chang EH. TP53 Gene Therapy as a Potential Treatment for Patients with COVID-19. Viruses. 2022; 14(4):739. https://doi.org/10.3390/v14040739
Chicago/Turabian StyleHarford, Joe B., Sang Soo Kim, Kathleen F. Pirollo, and Esther H. Chang. 2022. "TP53 Gene Therapy as a Potential Treatment for Patients with COVID-19" Viruses 14, no. 4: 739. https://doi.org/10.3390/v14040739