
KAP1 Positively Modulates Influenza A Virus Replication by Interacting with PB2 and NS1 Proteins in Human Lung Epithelial Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Viruses and Ethics
2.2. siRNA Transfection and Knock-Down Efficacy Detection
2.3. Generation of the KAP1 Overexpression and Knockout A549 Cells
2.4. Immunoprecipitation
2.5. Antibodies and Western Blotting
2.6. Virus Growth Kinetics
2.7. Immunofluorescence
2.8. PB2-KO/Rluc Virus Assay
2.9. Mini-Genome Assay
2.10. IFN-β ELISA
2.11. VLP Formation Assay
2.12. Single-Molecule Fluorescence In Situ Hybridization (smFISH)
2.13. Statistical Analysis
3. Results
3.1. KAP1 Is Required for Influenza A Replication in A549 Cells
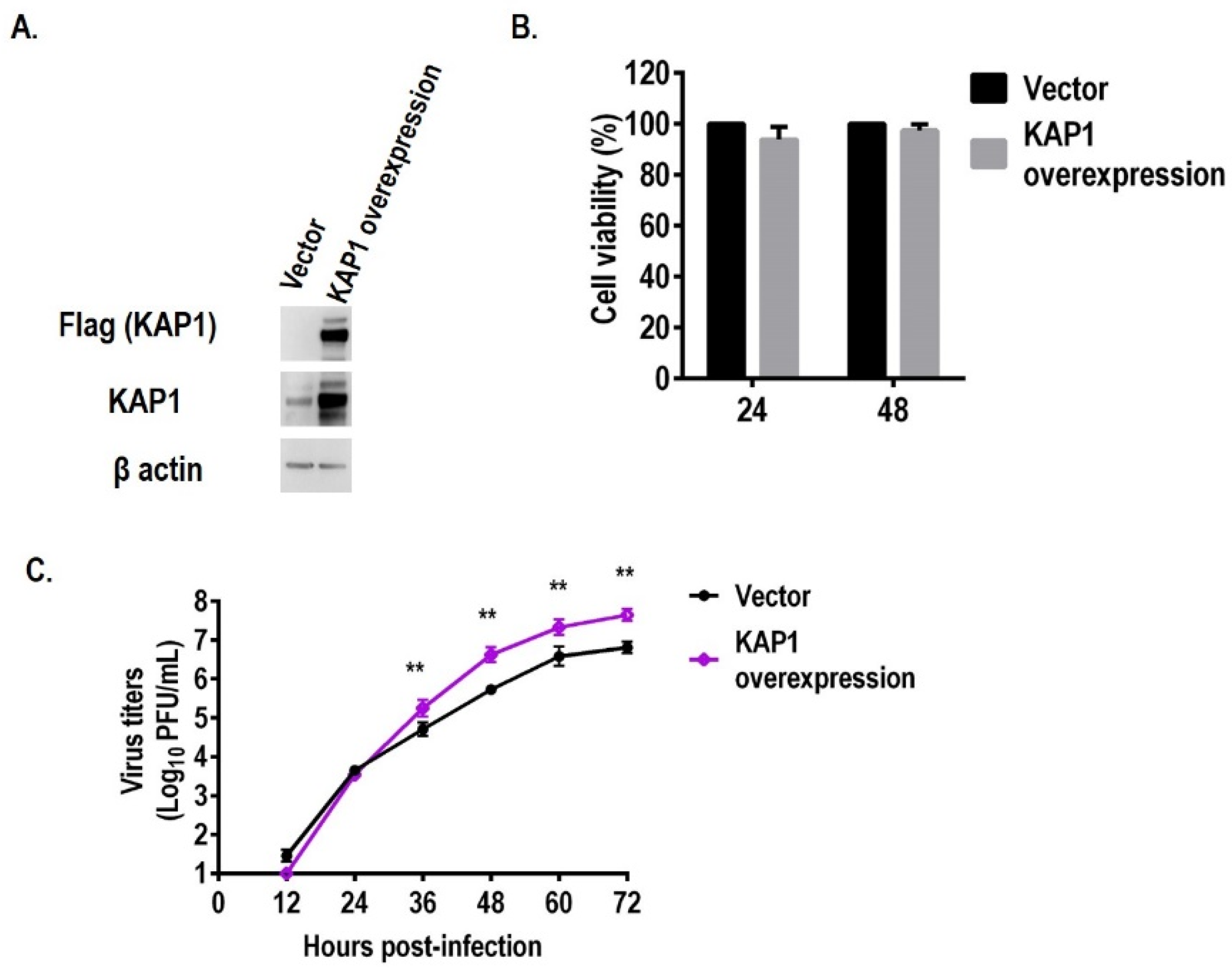
3.2. Overexpression of KAP1 Promotes the Replication of Influenza Virus in A549 Cells
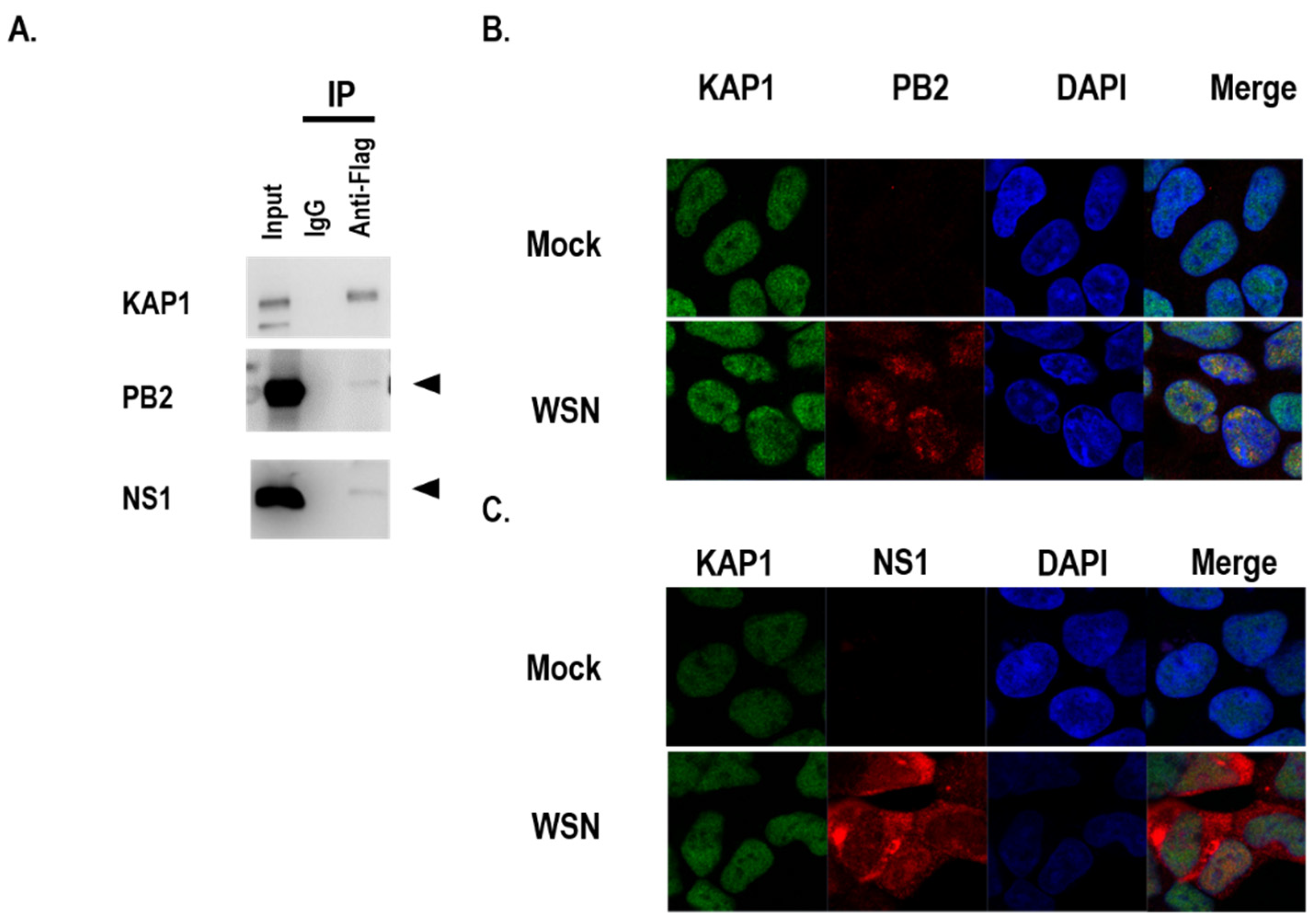
3.3. KAP1 Interacted with PB2 and NS1 Proteins
3.4. KAP1 Is Involved in the Early Steps of Influenza Virus Replication
3.5. Lack of KAP1 Reduced the Polymerase Activity of the Influenza A Virus
3.6. KAP1 Is Necessary for the Expression of Viral Proteins
3.7. KAP1 Negatively Regulated the Interferon Production after Influenza Virus Infection
3.8. KAP1 Was deSUMOylated and Phosphorylated during Influenza Infection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Han, J.; Perez, J.T.; Chen, C.; Li, Y.; Benitez, A.; Kandasamy, M.; Lee, Y.; Andrade, J.; tenOever, B.; Manicassamy, B. Genome-wide CRISPR/Cas9 Screen Identifies Host Factors Essential for Influenza Virus Replication. Cell Rep. 2018, 23, 596–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaton, B.E.; Kennedy, E.M.; Dumm, R.E.; Harding, A.T.; Sacco, M.T.; Sachs, D.; Heaton, N.S. A CRISPR Activation Screen Identifies a Pan-avian Influenza Virus Inhibitory Host Factor. Cell Rep. 2017, 20, 1503–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, T.; Kawakami, E.; Shoemaker, J.E.; Lopes, T.J.; Matsuoka, Y.; Tomita, Y.; Kozuka-Hata, H.; Gorai, T.; Kuwahara, T.; Takeda, E.; et al. Influenza Virus-Host Interactome Screen as a Platform for Antiviral Drug Development. Cell Host Microbe 2014, 16, 795–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.C.; Chen, Y.C.; Tseng, C.H.; Hsu, P.W.; Tung, K.F.; Jeng, K.S.; Lai, M.M. Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proc. Natl. Acad. Sci. USA 2013, 110, 17516–17521. [Google Scholar] [CrossRef] [Green Version]
- Konig, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef]
- Karlas, A.; Machuy, N.; Shin, Y.; Pleissner, K.P.; Artarini, A.; Heuer, D.; Becker, D.; Khalil, H.; Ogilvie, L.A.; Hess, S.; et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Shapira, S.D.; Gat-Viks, I.; Shum, B.O.; Dricot, A.; de Grace, M.M.; Wu, L.; Gupta, P.B.; Hao, T.; Silver, S.J.; Root, D.E.; et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 2009, 139, 1255–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, L.; Sakurai, A.; Watanabe, T.; Sorensen, E.; Nidom, C.A.; Newton, M.A.; Ahlquist, P.; Kawaoka, Y. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 2008, 454, 890–893. [Google Scholar] [CrossRef] [Green Version]
- Bradel-Tretheway, B.G.; Mattiacio, J.L.; Krasnoselsky, A.; Stevenson, C.; Purdy, D.; Dewhurst, S.; Katze, M.G. Comprehensive proteomic analysis of influenza virus polymerase complex reveals a novel association with mitochondrial proteins and RNA polymerase accessory factors. J. Virol. 2011, 85, 8569–8581. [Google Scholar] [CrossRef] [Green Version]
- Sripathy, S.P.; Stevens, J.; Schultz, D.C. The KAP1 corepressor functions to coordinate the assembly of de novo HP1-demarcated microenvironments of heterochromatin required for KRAB zinc finger protein-mediated transcriptional repression. Mol. Cell Biol. 2006, 26, 8623–8638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Short, K.M.; Cox, T.C. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J. Biol. Chem. 2006, 281, 8970–8980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 2019, 8, e42426. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Burton, E.M.; Bhaduri-McIntosh, S. Chloroquine triggers Epstein-Barr virus replication through phosphorylation of KAP1/TRIM28 in Burkitt lymphoma cells. PLoS Pathog. 2017, 13, e1006249. [Google Scholar] [CrossRef] [PubMed]
- Rauwel, B.; Jang, S.M.; Cassano, M.; Kapopoulou, A.; Barde, I.; Trono, D. Release of human cytomegalovirus from latency by a KAP1/TRIM28 phosphorylation switch. eLife 2015, 4, e06068. [Google Scholar] [CrossRef] [PubMed]
- Fasching, L.; Kapopoulou, A.; Sachdeva, R.; Petri, R.; Jonsson, M.E.; Manne, C.; Turelli, P.; Jern, P.; Cammas, F.; Trono, D.; et al. TRIM28 represses transcription of endogenous retroviruses in neural progenitor cells. Cell Rep. 2015, 10, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Burck, C.; Mund, A.; Berscheminski, J.; Kieweg, L.; Muncheberg, S.; Dobner, T.; Schreiner, S. KAP1 Is a Host Restriction Factor That Promotes Human Adenovirus E1B-55K SUMO Modification. J. Virol. 2015, 90, 930–946. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhu, C.; Guo, Y.; Wei, F.; Lu, J.; Qin, J.; Banerjee, S.; Wang, J.; Shang, H.; Verma, S.C.; et al. Inhibition of KAP1 Enhances Hypoxia-Induced Kaposi’s Sarcoma-Associated Herpesvirus Reactivation through RBP-Jkappa. J. Virol. 2014, 88, 6873–6884. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Liang, D.; Gao, Y.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded LANA interacts with host KAP1 to facilitate establishment of viral latency. J. Virol. 2014, 88, 7331–7344. [Google Scholar] [CrossRef] [Green Version]
- Allouch, A.; Di Primio, C.; Alpi, E.; Lusic, M.; Arosio, D.; Giacca, M.; Cereseto, A. The TRIM family protein KAP1 inhibits HIV-1 integration. Cell Host Microbe 2011, 9, 484–495. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Fan, Y.; Huang, Y.; Du, T.; Liu, Z.; Huang, D.; Wang, Y.; Wang, N.; Zhang, P. TRIM28 regulates SARS-CoV-2 cell entry by targeting ACE2. Cell Signal 2021, 85, 110064. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Moshkina, N.; Fenouil, R.; Gardner, T.J.; Aguirre, S.; Shah, P.S.; Zhao, N.; Manganaro, L.; Hultquist, J.F.; Noel, J.; et al. Targeting Viral Proteostasis Limits Influenza Virus, HIV, and Dengue Virus Infection. Immunity 2016, 44, 46–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dapat, C.; Saito, R.; Suzuki, H.; Horigome, T. Quantitative phosphoproteomic analysis of host responses in human lung epithelial (A549) cells during influenza virus infection. Virus Res. 2014, 179, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.; Watanabe, T.; Ito, H.; Watanabe, S.; Goto, H.; Gao, P.; Hughes, M.; Perez, D.R.; Donis, R.; Hoffmann, E.; et al. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. USA 1999, 96, 9345–9350. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, M.; Victor, S.T.; Taft, A.S.; Yamada, S.; Li, C.; Hatta, M.; Das, S.C.; Takashita, E.; Kakugawa, S.; Maher, E.A.; et al. Replication-incompetent influenza A viruses that stably express a foreign gene. J. Gen. Virol. 2011, 92, 2879–2888. [Google Scholar] [CrossRef]
- Octaviani, C.P.; Ozawa, M.; Yamada, S.; Goto, H.; Kawaoka, Y. High level of genetic compatibility between swine-origin H1N1 and highly pathogenic avian H5N1 influenza viruses. J. Virol. 2010, 84, 10918–10922. [Google Scholar] [CrossRef] [Green Version]
- Daniels, R.S.; Downie, J.C.; Hay, A.J.; Knossow, M.; Skehel, J.J.; Wang, M.L.; Wiley, D.C. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 1985, 40, 431–439. [Google Scholar] [CrossRef]
- Bunch, H.; Calderwood, S.K. TRIM28 as a novel transcriptional elongation factor. BMC Mol. Biol. 2015, 16, 14. [Google Scholar] [CrossRef] [Green Version]
- Bunch, H.; Zheng, X.; Burkholder, A.; Dillon, S.T.; Motola, S.; Birrane, G.; Ebmeier, C.C.; Levine, S.; Fargo, D.; Hu, G.; et al. TRIM28 regulates RNA polymerase II promoter-proximal pausing and pause release. Nat. Struct. Mol. Biol. 2014, 21, 876–883. [Google Scholar] [CrossRef] [Green Version]
- Patil, G.; Li, S.T. Tripartite motif proteins: An emerging antiviral protein family. Future Virol. 2019, 14, 107–122. [Google Scholar] [CrossRef]
- Watanabe, M.; Hatakeyama, S. TRIM proteins and diseases. J. Biochem. 2017, 161, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Van Tol, S.; Hage, A.; Giraldo, M.I.; Bharaj, P.; Rajsbaum, R. The TRIMendous Role of TRIMs in Virus-Host Interactions. Vaccines 2017, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajsbaum, R.; Stoye, J.P.; O’Garra, A. Type I interferon-dependent and -independent expression of tripartite motif proteins in immune cells. Eur. J. Immunol. 2008, 38, 619–630. [Google Scholar] [CrossRef]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C., 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayllon, J.; Garcia-Sastre, A. The NS1 protein: A multitasking virulence factor. Curr. Top. Microbiol. Immunol. 2015, 386, 73–107. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Donelan, N.R.; Basler, C.F.; Garcia-Sastre, A. A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J. Virol. 2003, 77, 13257–13266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talon, J.; Horvath, C.M.; Polley, R.; Basler, C.F.; Muster, T.; Palese, P.; Garcia-Sastre, A. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J. Virol. 2000, 74, 7989–7996. [Google Scholar] [CrossRef] [Green Version]
- Patil, G.; Zhao, M.; Song, K.; Hao, W.; Bouchereau, D.; Wang, L.; Li, S. TRIM41-Mediated Ubiquitination of Nucleoprotein Limits Influenza A Virus Infection. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Meyerson, N.R.; Zhou, L.; Guo, Y.R.; Zhao, C.; Tao, Y.J.; Krug, R.M.; Sawyer, S.L. Nuclear TRIM25 Specifically Targets Influenza Virus Ribonucleoproteins to Block the Onset of RNA Chain Elongation. Cell Host Microbe 2017, 22, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Li, N.L.; Shen, Y.; Bao, X.; Fabrizio, T.; Elbahesh, H.; Webby, R.J.; Li, K. The C-terminal Tail of TRIM56 Dictates Antiviral Restriction of Influenza A and B Viruses by Impeding Viral RNA Synthesis. J. Virol. 2016, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, B.; Wang, L.; Ding, H.; Schwamborn, J.C.; Li, S.; Dorf, M.E. TRIM32 Senses and Restricts Influenza A Virus by Ubiquitination of PB1 Polymerase. PLoS Pathog. 2015, 11, e1004960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 2013, 87, 4523–4533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Zhang, J.; Xue, Q.; Liu, J.; Huang, B.; He, Z.; Huang, J.; Zu, S.; Chen, Z.; Zhao, B.; et al. Duck TRIM32 Functions in IFN-beta Signaling Against the Infection of H5N6 Highly Pathogenic Avian Influenza Virus. Front. Immunol. 2020, 11, 377. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Jiang, L.; Ye, M.; Wang, Y.; Wang, G.; Wan, X.; Zhao, Y.; Wen, X.; Liang, L.; Ma, S.; et al. TRIM35 mediates protection against influenza infection by activating TRAF3 and degrading viral PB2. Protein Cell 2020, 11, 894–914. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Weng, L.; Yuan, B.; Wang, Z.; Jia, L.; Jin, R.; Lu, H.; Li, X.C.; Liu, Y.J.; Zhang, Z. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat. Immunol. 2016, 17, 1373–1380. [Google Scholar] [CrossRef]
- Kochs, G.; Garcia-Sastre, A.; Martinez-Sobrido, L. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 2007, 81, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef]
- Chen, G.; Liu, C.H.; Zhou, L.; Krug, R.M. Cellular DDX21 RNA helicase inhibits influenza A virus replication but is counteracted by the viral NS1 protein. Cell Host Microbe 2014, 15, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, N.; Domingues, P.; Golebiowski, F.; Patzina, C.; Tatham, M.H.; Hay, R.T.; Hale, B.G. An influenza virus-triggered SUMO switch orchestrates co-opted endogenous retroviruses to stimulate host antiviral immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 17399–17408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krischuns, T.; Gunl, F.; Henschel, L.; Binder, M.; Willemsen, J.; Schloer, S.; Rescher, U.; Gerlt, V.; Zimmer, G.; Nordhoff, C.; et al. Phosphorylation of TRIM28 Enhances the Expression of IFN-beta and Proinflammatory Cytokines During HPAIV Infection of Human Lung Epithelial Cells. Front. Immunol. 2018, 9, 2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Klenk, C.; Liu, B.; Keiner, B.; Cheng, J.; Zheng, B.J.; Li, L.; Han, Q.; Wang, C.; Li, T.; et al. Modification of nonstructural protein 1 of influenza A virus by SUMO1. J. Virol. 2011, 85, 1086–1098. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.; Pal, S.; Chacon, J.; Meraz, K.; Gonzalez, J.; Prieto, K.; Rosas-Acosta, G. SUMOylation affects the interferon blocking activity of the influenza A nonstructural protein NS1 without affecting its stability or cellular localization. J. Virol. 2013, 87, 5602–5620. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, H.; Yi, R.; Wu, S.; Wang, G.; Sun, R.; Lin, L.; Zhu, S.; Nie, Z.; He, Y.; Wang, S.; et al. KAP1 Positively Modulates Influenza A Virus Replication by Interacting with PB2 and NS1 Proteins in Human Lung Epithelial Cells. Viruses 2022, 14, 689. https://doi.org/10.3390/v14040689
Feng H, Yi R, Wu S, Wang G, Sun R, Lin L, Zhu S, Nie Z, He Y, Wang S, et al. KAP1 Positively Modulates Influenza A Virus Replication by Interacting with PB2 and NS1 Proteins in Human Lung Epithelial Cells. Viruses. 2022; 14(4):689. https://doi.org/10.3390/v14040689
Chicago/Turabian StyleFeng, Huapeng, Ruonan Yi, Shixiang Wu, Genzhu Wang, Ruolin Sun, Liming Lin, Shunfan Zhu, Zhenyu Nie, Yulong He, Siquan Wang, and et al. 2022. "KAP1 Positively Modulates Influenza A Virus Replication by Interacting with PB2 and NS1 Proteins in Human Lung Epithelial Cells" Viruses 14, no. 4: 689. https://doi.org/10.3390/v14040689