
The Interplay between Viruses and Host DNA Sensors


Abstract
:1. Introduction
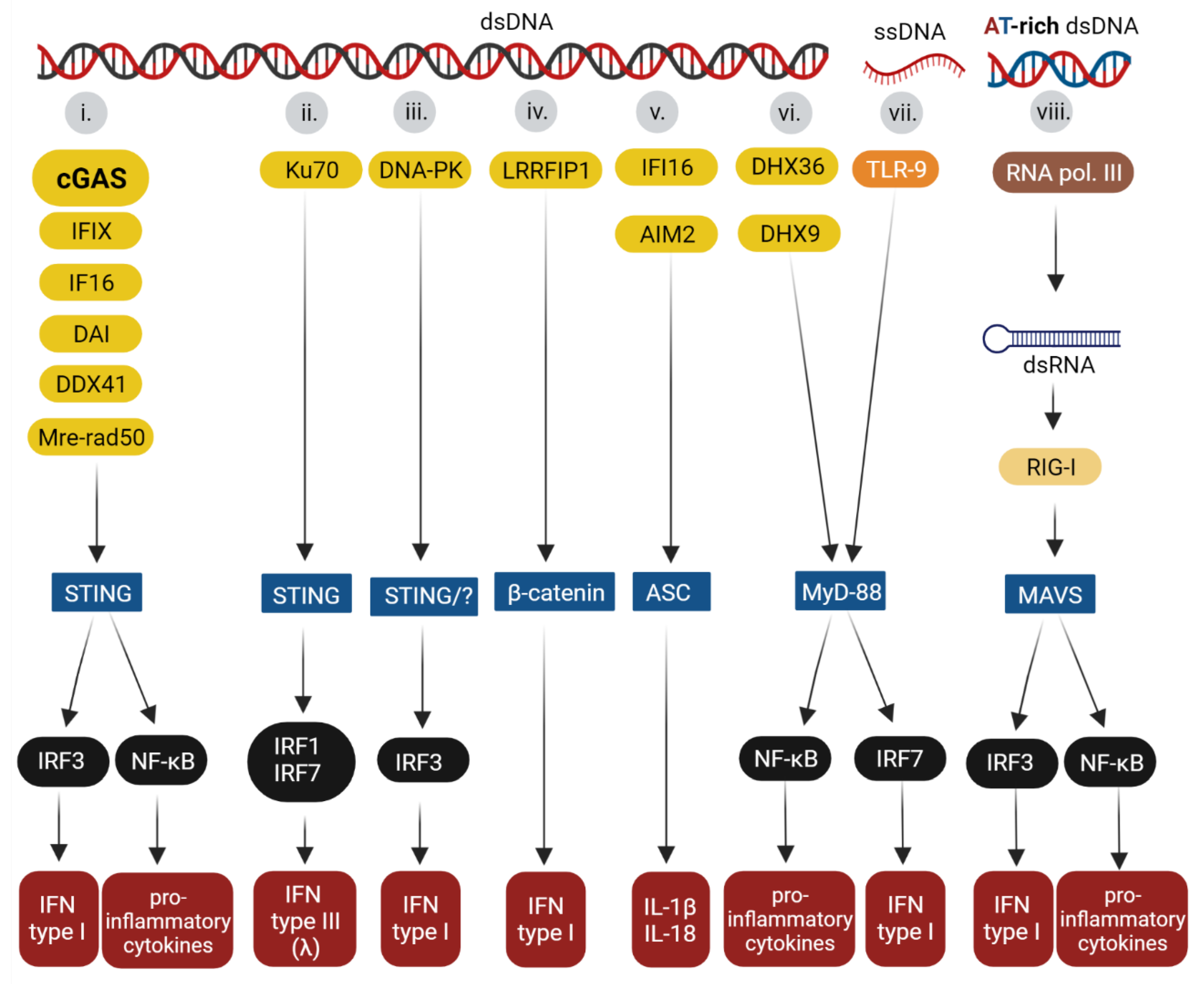
2. Overview of the Innate Immune Sensors
3. Molecular Bases of DNA Recognition
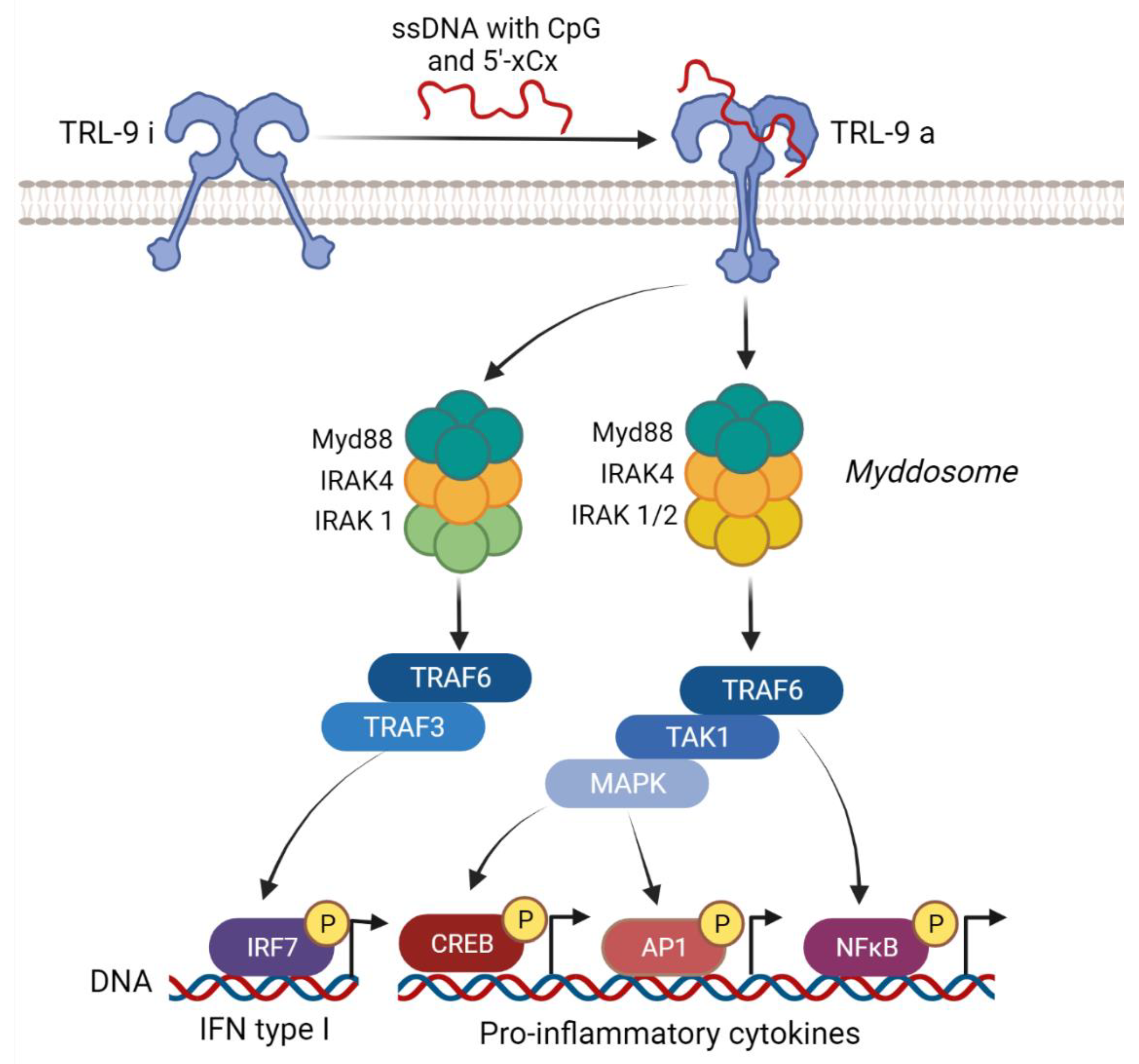
3.1. TLR9 Signaling Pathway
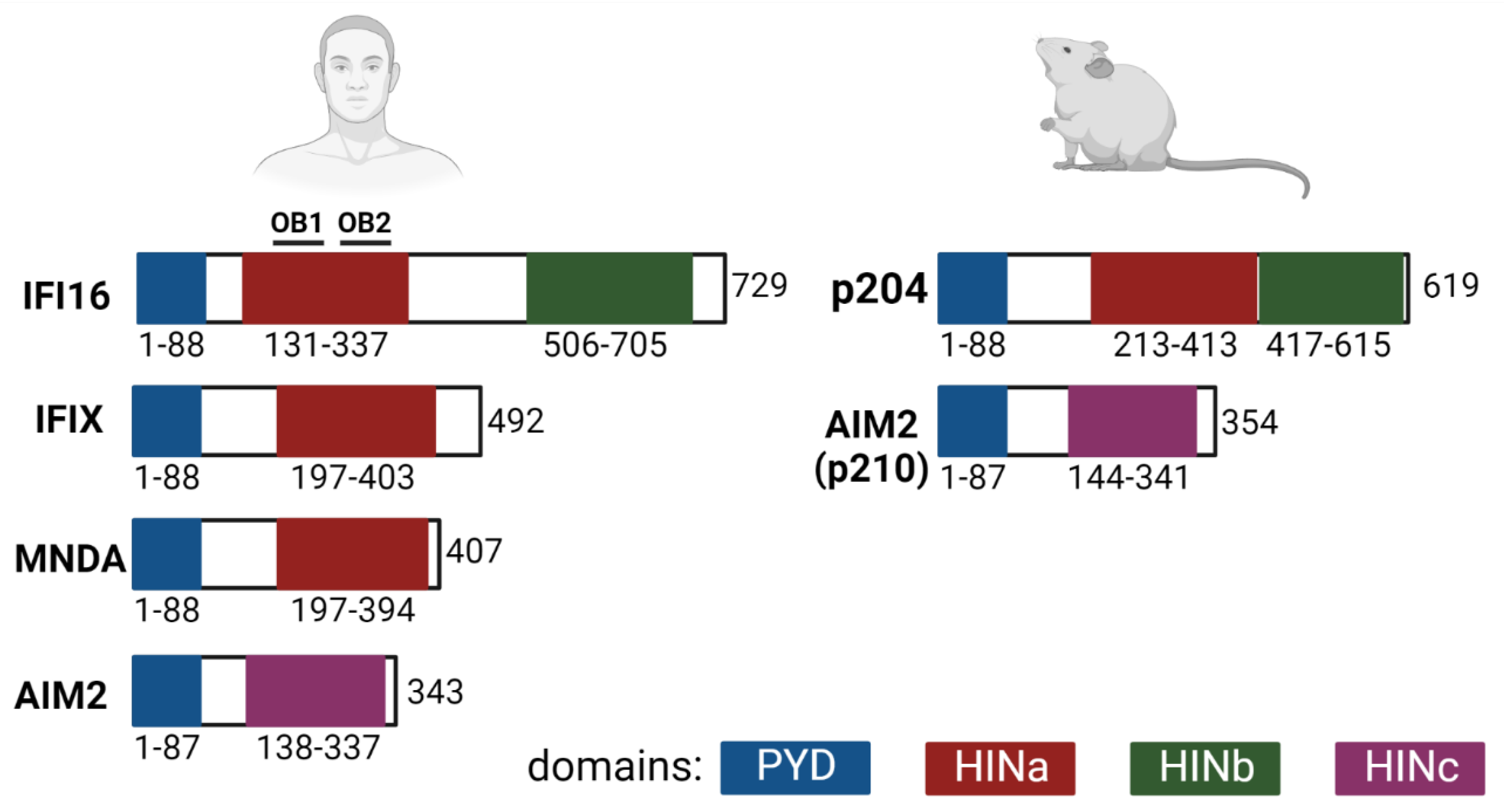
3.2. The PYHIN Family Proteins IFI16, p204/Ifi-204 and AIM2: The Inflammasome and the IFN Signaling Pathways
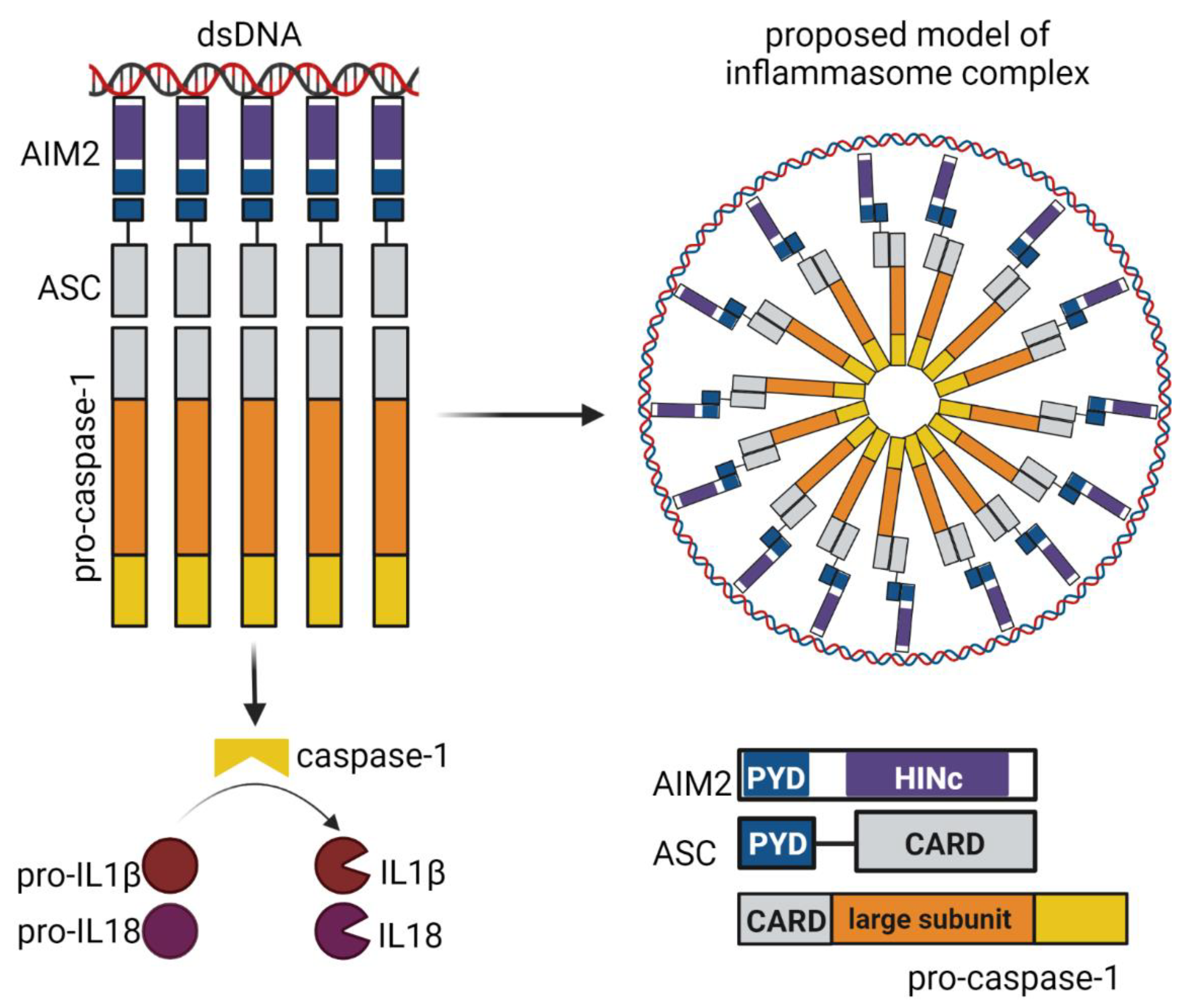
3.2.1. AIM2 Inflammasome Signaling Pathway
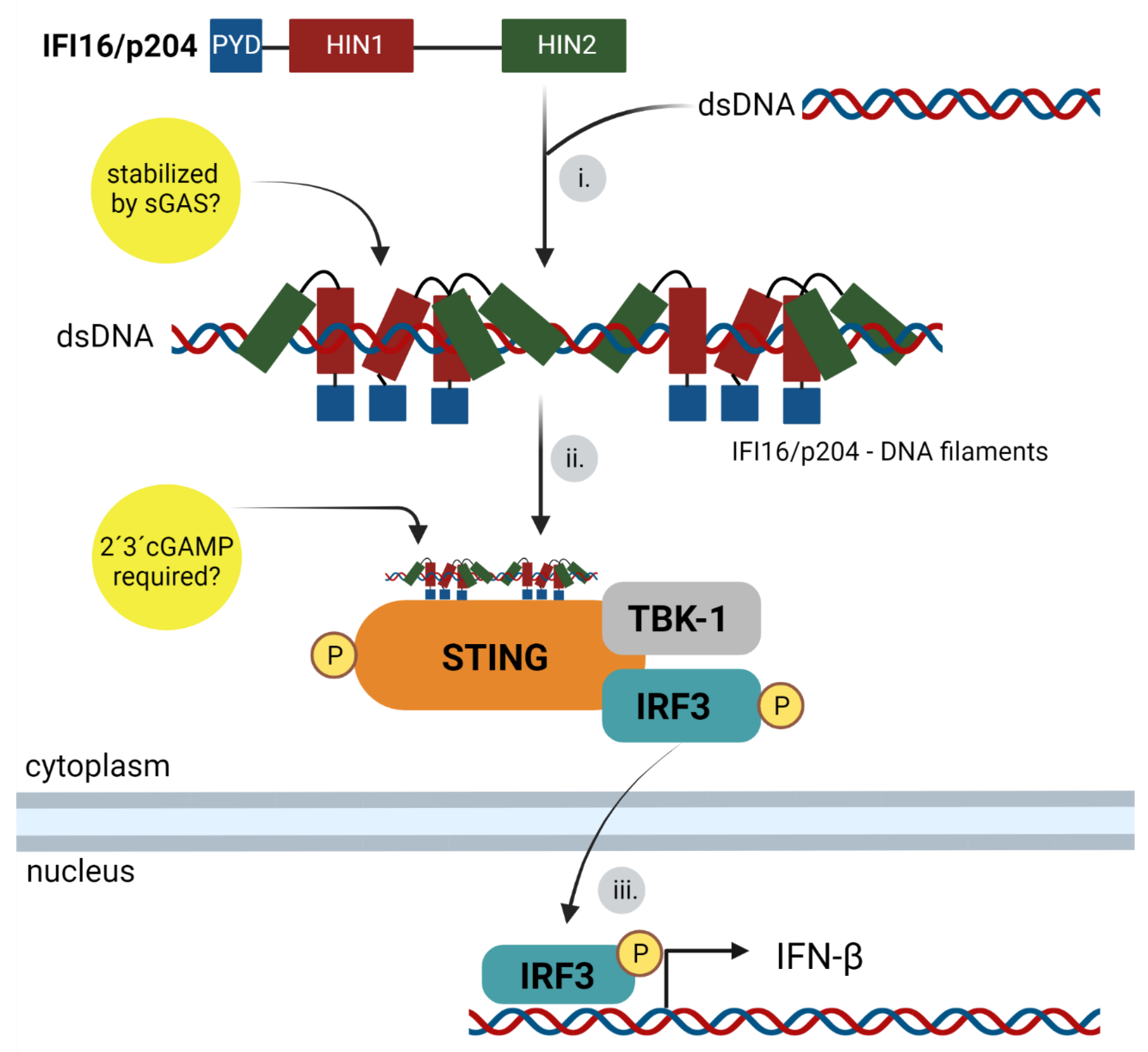
3.2.2. IFI16 and p204/Ifi-204 Binding to DNA and IFN Signaling Pathway
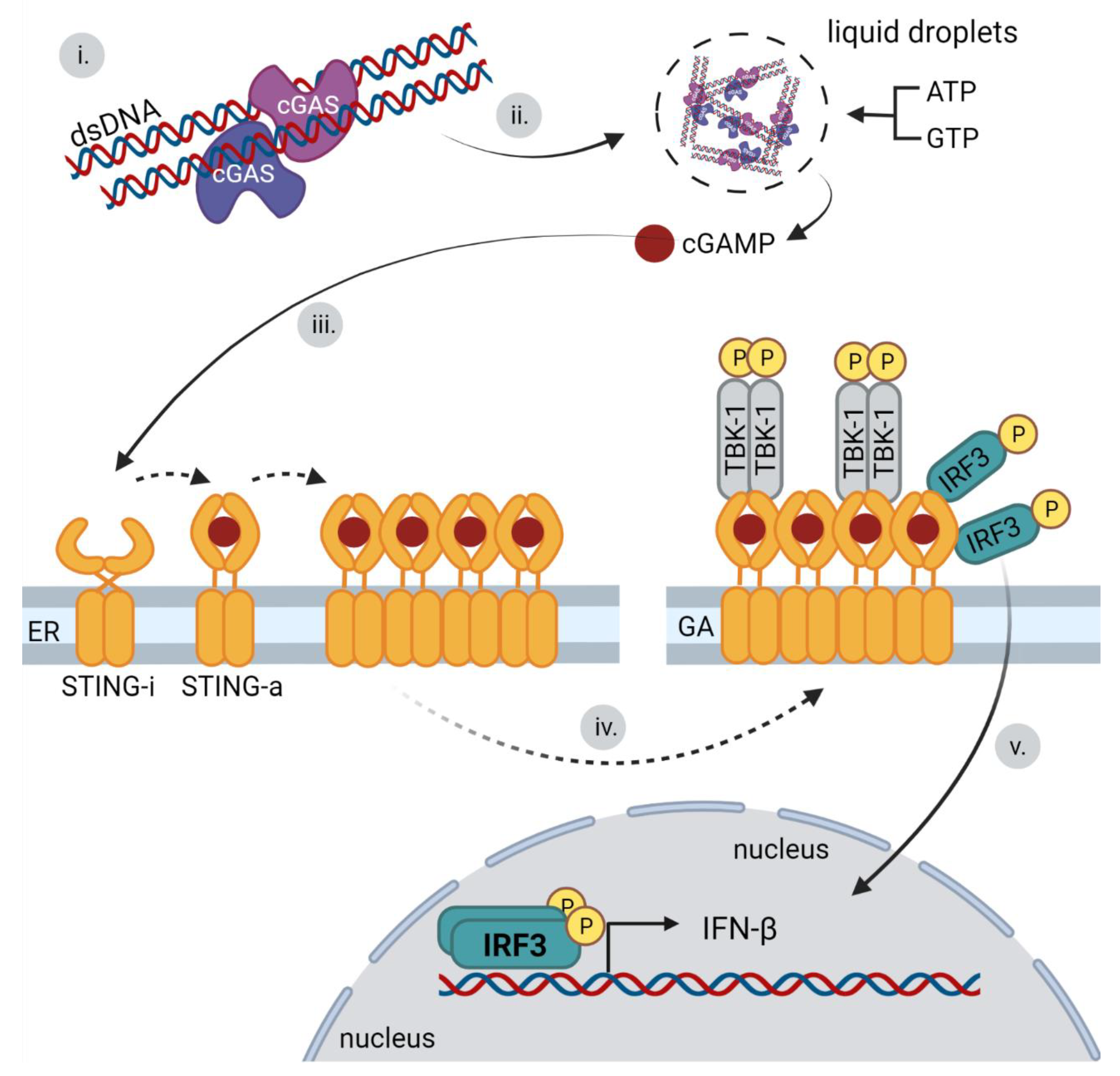
3.3. cGAS Binding to DNA and IFN Signaling Pathway
4. Sensing of Viral Genomes by DNA Sensors
4.1. Immune Sensing of Human Herpesvirus Genomes
4.2. Adenoviruses Sensing by DNA Sensors
4.3. Interplay between DNA Sensors and Human Papillomaviruses
4.4. Polyomaviruses Interactions with DNA Immune Sensing Pathways
4.5. HBV Virus and the DNA Innate Immune Sensing Pathways
4.6. HIV and DNA Innate Immune Sensing Pathways
5. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saxena, M.; Yeretssian, G. NOD-Like Receptors: Master Regulators of Inflammation and Cancer. Front. Immunol. 2014, 5, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drouin, M.; Saenz, J.; Chiffoleau, E. C-Type Lectin-Like Receptors: Head or Tail in Cell Death Immunity. Front. Immunol. 2020, 11, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. The Roles of TLRs, RLRs and NLRs in Pathogen Recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira Mann, C.C.; Hornung, V. Molecular Mechanisms of Nonself Nucleic Acid Recognition by the Innate Immune System. Eur. J. Immunol. 2021, 51, 1897–1910. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like Receptors: Their Regulation and Roles in RNA Sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Caneparo, V.; Landolfo, S.; Gariglio, M.; De Andrea, M. The Absent in Melanoma 2-Like Receptor IFN-Inducible Protein 16 as an Inflammasome Regulator in Systemic Lupus Erythematosus: The Dark Side of Sensing Microbes. Front. Immunol. 2018, 9, 1180. [Google Scholar] [CrossRef]
- Bürckstümmer, T.; Baumann, C.; Blüml, S.; Dixit, E.; Dürnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An Orthogonal Proteomic-Genomic Screen Identifies AIM2 as a Cytoplasmic DNA Sensor for the Inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef]
- Howard, T.R.; Crow, M.S.; Greco, T.M.; Lum, K.K.; Li, T.; Cristea, I.M. The DNA Sensor IFIX Drives Proteome Alterations To Mobilize Nuclear and Cytoplasmic Antiviral Responses, with Its Acetylation Acting as a Localization Toggle. mSystems 2021, 6, e00397-21. [Google Scholar] [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 Is an Innate Immune Sensor for Intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Diner, B.A.; Li, T.; Greco, T.M.; Crow, M.S.; Fuesler, J.A.; Wang, J.; Cristea, I.M. The Functional Interactome of PYHIN Immune Regulators Reveals IFIX Is a Sensor of Viral DNA. Mol. Syst. Biol. 2015, 11, 787. [Google Scholar] [CrossRef]
- Chen, W.; Yu, S.-X.; Zhou, F.-H.; Zhang, X.-J.; Gao, W.-Y.; Li, K.-Y.; Liu, Z.-Z.; Han, W.-Y.; Yang, Y.-J. DNA Sensor IFI204 Contributes to Host Defense Against Staphylococcus Aureus Infection in Mice. Front. Immunol. 2019, 10, 474. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) Is a Cytosolic DNA Sensor and an Activator of Innate Immune Response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.-J. The Helicase DDX41 Senses Intracellular DNA Mediated by the Adaptor STING in Dendritic Cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.; Pazhoor, S.; Bao, M.; Zhang, Z.; Hanabuchi, S.; Facchinetti, V.; Bover, L.; Plumas, J.; Chaperot, L.; Qin, J.; et al. Aspartate-Glutamate-Alanine-Histidine Box Motif (DEAH)/RNA Helicase A Helicases Sense Microbial DNA in Human Plasmacytoid Dendritic Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 15181–15186. [Google Scholar] [CrossRef] [Green Version]
- Ng, Y.C.; Chung, W.-C.; Kang, H.-R.; Cho, H.-J.; Park, E.-B.; Kang, S.-J.; Song, M.J. A DNA-Sensing–Independent Role of a Nuclear RNA Helicase, DHX9, in Stimulation of NF-ΚB–Mediated Innate Immunity against DNA Virus Infection. Nucleic Acids Res. 2018, 46, 9011–9026. [Google Scholar] [CrossRef]
- Chiu, Y.-H.; MacMillan, J.B.; Chen, Z.J. RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Kobayashi, J.; Saitoh, T.; Maruyama, K.; Ishii, K.J.; Barber, G.N.; Komatsu, K.; Akira, S.; Kawai, T. DNA Damage Sensor MRE11 Recognizes Cytosolic Double-Stranded DNA and Induces Type I Interferon by Regulating STING Trafficking. Proc. Natl. Acad. Sci. USA 2013, 110, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK Is a DNA Sensor for IRF-3-Dependent Innate Immunity. eLife 2012, 1, e00047. [Google Scholar] [CrossRef]
- Sui, H.; Hao, M.; Chang, W.; Imamichi, T. The Role of Ku70 as a Cytosolic DNA Sensor in Innate Immunity and Beyond. Front. Cell. Infect. Microbiol. 2021, 11, 761983. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Wei, W.; Greenway, A.; Trapani, J.A. Functional Interaction between P53 and the Interferon-Inducible Nucleoprotein IFI 16. Oncogene 2000, 19, 6033–6042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, R.W.; Kerry, J.A.; Trapani, J.A. The Human Interferon-Inducible Protein, IFI 16, Is a Repressor of Transcription. J. Biol. Chem. 1998, 273, 17172–17177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Ghosh, A.; Kumar, B.; Chandran, B. IFI16, a Nuclear Innate Immune DNA Sensor, Mediates Epigenetic Silencing of Herpesvirus Genomes by Its Association with H3K9 Methyltransferases SUV39H1 and GLP. eLife 2019, 8, e49500. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Bottero, V.; Flaherty, S.; Dutta, S.; Singh, V.V.; Chandran, B. IFI16 Restricts HSV-1 Replication by Accumulating on the HSV-1 Genome, Repressing HSV-1 Gene Expression, and Directly or Indirectly Modulating Histone Modifications. PLoS Pathog. 2014, 10, e1004503. [Google Scholar] [CrossRef]
- Jiang, H.; Xue, X.; Panda, S.; Kawale, A.; Hooy, R.M.; Liang, F.; Sohn, J.; Sung, P.; Gekara, N.O. Chromatin-bound cGAS Is an Inhibitor of DNA Repair and Hence Accelerates Genome Destabilization and Cell Death. EMBO J. 2019, 38, e102718. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear CGAS Suppresses DNA Repair and Promotes Tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef]
- Zierhut, C.; Yamaguchi, N.; Paredes, M.; Luo, J.-D.; Carroll, T.; Funabiki, H. The Cytoplasmic DNA Sensor CGAS Promotes Mitotic Cell Death. Cell 2019, 178, 302–315.e23. [Google Scholar] [CrossRef]
- Cui, S.; Yu, Q.; Chu, L.; Cui, Y.; Ding, M.; Wang, Q.; Wang, H.; Chen, Y.; Liu, X.; Wang, C. Nuclear CGAS Functions Non-Canonically to Enhance Antiviral Immunity via Recruiting Methyltransferase Prmt5. Cell Rep. 2020, 33, 108490. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. CGAS Produces a 2′-5′-Linked Cyclic Dinucleotide Second Messenger That Activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Sui, H.; Zhou, M.; Imamichi, H.; Jiao, X.; Sherman, B.T.; Lane, H.C.; Imamichi, T. STING Is an Essential Mediator of the Ku70-Mediated Production of IFN-Λ1 in Response to Exogenous DNA. Sci. Signal. 2017, 10, eaah5054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burleigh, K.; Maltbaek, J.H.; Cambier, S.; Green, R.; Gale, M.; James, R.C.; Stetson, D.B. Human DNA-PK Activates a STING-Independent DNA Sensing Pathway. Sci. Immunol. 2020, 5, eaba4219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; An, H.; Liu, X.; Wen, M.; Zheng, Y.; Rui, Y.; Cao, X. The Cytosolic Nucleic Acid Sensor LRRFIP1 Mediates the Production of Type I Interferon via a β-Catenin-Dependent Pathway. Nat. Immunol. 2010, 11, 487–494. [Google Scholar] [CrossRef]
- Liu, Y.; Zou, Z.; Zhu, B.; Hu, Z.; Zeng, P.; Wu, L. LRRFIP1 Inhibits Hepatitis C Virus Replication by Inducing Type I Interferon in Hepatocytes. Hepat. Mon. 2015, 15, e28473. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.K.; Sharma, S.; Fitzgerald, K.A. Catenin’ on to Nucleic Acid Sensing. Nat. Immunol. 2010, 11, 466–468. [Google Scholar] [CrossRef]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like Receptor Recognizes Bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, N.; Ho, S.; Antonenko, S.; de Waal Malefyt, R.; Kastelein, R.A.; Bazan, F.; Liu, Y.-J. Subsets of Human Dendritic Cell Precursors Express Different Toll-like Receptors and Respond to Different Microbial Antigens. J. Exp. Med. 2001, 194, 863–870. [Google Scholar] [CrossRef]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdörfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative Expression of Toll-Like Receptor 1–10 MRNA in Cellular Subsets of Human Peripheral Blood Mononuclear Cells and Sensitivity to CpG Oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [Green Version]
- Botos, I.; Segal, D.M.; Davies, D.R. The Structural Biology of Toll-like Receptors. Structure 2011, 19, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Avalos, A.M.; Kirak, O.; Oelkers, J.M.; Pils, M.C.; Kim, Y.-M.; Ottinger, M.; Jaenisch, R.; Ploegh, H.L.; Brinkmann, M.M. Cell-Specific TLR9 Trafficking in Primary APCs of Transgenic TLR9-GFP Mice. J. Immunol. 2013, 190, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Chockalingam, A.; Brooks, J.C.; Cameron, J.L.; Blum, L.K.; Leifer, C.A. TLR9 Traffics through the Golgi Complex to Localize to Endolysosomes and Respond to CpG DNA. Immunol. Cell Biol. 2009, 87, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, J.C.; Barton, G.M. Emerging Principles Governing Signal Transduction by Pattern-Recognition Receptors. Cold Spring Harb. Perspect. Biol. 2014, 7, a016253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like Receptors and Innate Immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Ohto, U.; Ishida, H.; Shibata, T.; Sato, R.; Miyake, K.; Shimizu, T. Toll-like Receptor 9 Contains Two DNA Binding Sites That Function Cooperatively to Promote Receptor Dimerization and Activation. Immunity 2018, 48, 649–658.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latz, E.; Verma, A.; Visintin, A.; Gong, M.; Sirois, C.M.; Klein, D.C.G.; Monks, B.G.; McKnight, C.J.; Lamphier, M.S.; Duprex, W.P.; et al. Ligand-Induced Conformational Changes Allosterically Activate Toll-like Receptor 9. Nat. Immunol. 2007, 8, 772–779. [Google Scholar] [CrossRef]
- Häcker, H.; Redecke, V.; Blagoev, B.; Kratchmarova, I.; Hsu, L.-C.; Wang, G.G.; Kamps, M.P.; Raz, E.; Wagner, H.; Häcker, G.; et al. Specificity in Toll-like Receptor Signalling through Distinct Effector Functions of TRAF3 and TRAF6. Nature 2006, 439, 204–207. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; et al. Interferon-α Induction through Toll-like Receptors Involves a Direct Interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef]
- Marongiu, L.; Gornati, L.; Artuso, I.; Zanoni, I.; Granucci, F. Below the Surface: The Inner Lives of TLR4 and TLR9. J. Leukoc. Biol. 2019, 106, 147–160. [Google Scholar] [CrossRef]
- Hoshino, K.; Sasaki, I.; Sugiyama, T.; Yano, T.; Yamazaki, C.; Yasui, T.; Kikutani, H.; Kaisho, T. Cutting Edge: Critical Role of IκB Kinase α in TLR7/9-Induced Type I IFN Production by Conventional Dendritic Cells. J.I. 2010, 184, 3341–3345. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, F.; Heit, A.; Guggemoos, S.; Krug, A.; Mages, J.; Schiemann, M.; Adler, H.; Drexler, I.; Haas, T.; Lang, R.; et al. Interferon-Regulatory-Factor 1 Controls Toll-like Receptor 9-Mediated IFN-β Production in Myeloid Dendritic Cells. Eur. J. Immunol. 2007, 37, 315–327. [Google Scholar] [CrossRef]
- Cridland, J.A.; Curley, E.Z.; Wykes, M.N.; Schroder, K.; Sweet, M.J.; Roberts, T.L.; Ragan, M.A.; Kassahn, K.S.; Stacey, K.J. The Mammalian PYHIN Gene Family: Phylogeny, Evolution and Expression. BMC Evol. Biol. 2012, 12, 140. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Gonzalezgugel, E.; Cheng, L.; Richbourgh, B.; Nie, L.; Liu, C. The Roles of Interferon-Inducible P200 Family Members IFI16 and P204 in Innate Immune Responses, Cell Differentiation and Proliferation. Genes Dis. 2015, 2, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, E.E.; Winship, D.; Snyder, J.M.; Child, S.J.; Geballe, A.P.; Stetson, D.B. The AIM2-like Receptors Are Dispensable for the Interferon Response to Intracellular DNA. Immunity 2016, 45, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakaya, Y.; Lilue, J.; Stavrou, S.; Moran, E.A.; Ross, S.R. AIM2-Like Receptors Positively and Negatively Regulate the Interferon Response Induced by Cytosolic DNA. mBio 2017, 8, e00944-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, N.; Liu, Z.-J. Role of the HIN Domain in Regulation of Innate Immune Responses. Mol. Cell. Biol. 2014, 34, 2–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrone, S.R.; Wang, T.; Constantoulakis, L.M.; Hooy, R.M.; Delannoy, M.J.; Sohn, J. Cooperative Assembly of IFI16 Filaments on DsDNA Provides Insights into Host Defense Strategy. Proc. Natl. Acad. Sci. USA 2014, 111, E62–E71. [Google Scholar] [CrossRef] [Green Version]
- Jin, T.; Perry, A.; Jiang, J.; Smith, P.; Curry, J.A.; Unterholzner, L.; Jiang, Z.; Horvath, G.; Rathinam, V.A.; Johnstone, R.W.; et al. Structures of the HIN Domain:DNA Complexes Reveal Ligand Binding and Activation Mechanisms of the AIM2 Inflammasome and IFI16 Receptor. Immunity 2012, 36, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Jin, T.; Perry, A.; Smith, P.; Jiang, J.; Xiao, T.S. Structure of the Absent in Melanoma 2 (AIM2) Pyrin Domain Provides Insights into the Mechanisms of AIM2 Autoinhibition and Inflammasome Assembly. J. Biol. Chem. 2013, 288, 13225–13235. [Google Scholar] [CrossRef] [Green Version]
- Morrone, S.R.; Matyszewski, M.; Yu, X.; Delannoy, M.; Egelman, E.H.; Sohn, J. Assembly-Driven Activation of the AIM2 Foreign-DsDNA Sensor Provides a Polymerization Template for Downstream ASC. Nat. Commun. 2015, 6, 7827. [Google Scholar] [CrossRef]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [Green Version]
- Ni, X.; Ru, H.; Ma, F.; Zhao, L.; Shaw, N.; Feng, Y.; Ding, W.; Gong, W.; Wang, Q.; Ouyang, S.; et al. New Insights into the Structural Basis of DNA Recognition by HINa and HINb Domains of IFI16. J. Mol. Cell Biol. 2016, 8, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Jiang, J.; Zhao, D.; Chen, F.; Ma, H.; Smith, P.; Unterholzner, L.; Xiao, T.S.; Jin, T. Structural Mechanism of DNA Recognition by the P204 HIN Domain. Nucleic Acids Res. 2021, 49, 2959–2972. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING Regulates Intracellular DNA-Mediated, Type I Interferon-Dependent Innate Immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Chen, Z.J. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci. Signal. 2012, 5, ra20. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.; Chen, Z.J. Structural Basis of STING Binding with and Phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.-Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The Adaptor Protein MITA Links Virus-Sensing Receptors to IRF3 Transcription Factor Activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [Green Version]
- Parker, Z.M.; Murphy, A.A.; Leib, D.A. Role of the DNA Sensor STING in Protection from Lethal Infection Following Corneal and Intracerebral Challenge with Herpes Simplex Virus 1. J. Virol. 2015, 89, 11080–11091. [Google Scholar] [CrossRef] [Green Version]
- Reinert, L.S.; Lopušná, K.; Winther, H.; Sun, C.; Thomsen, M.K.; Nandakumar, R.; Mogensen, T.H.; Meyer, M.; Vægter, C.; Nyengaard, J.R.; et al. Sensing of HSV-1 by the CGAS–STING Pathway in Microglia Orchestrates Antiviral Defence in the CNS. Nat. Commun. 2016, 7, 13348. [Google Scholar] [CrossRef] [Green Version]
- Stetson, D.B.; Medzhitov, R. Recognition of Cytosolic DNA Activates an IRF3-Dependent Innate Immune Response. Immunity 2006, 24, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Stratmann, S.A.; Morrone, S.R.; van Oijen, A.M.; Sohn, J. The Innate Immune Sensor IFI16 Recognizes Foreign DNA in the Nucleus by Scanning along the Duplex. eLife 2015, 4, e11721. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Dutta, S.; Veettil, M.V.; Dutta, D.; Iqbal, J.; Kumar, B.; Roy, A.; Chikoti, L.; Singh, V.V.; Chandran, B. Herpesvirus Genome Recognition Induced Acetylation of Nuclear IFI16 Is Essential for Its Cytoplasmic Translocation, Inflammasome and IFN-β Responses. PLoS Pathog. 2015, 11, e1005019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lum, K.K.; Howard, T.R.; Pan, C.; Cristea, I.M. Charge-Mediated Pyrin Oligomerization Nucleates Antiviral IFI16 Sensing of Herpesvirus DNA. mBio 2019, 10, e01428-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orzalli, M.H.; Broekema, N.M.; Diner, B.A.; Hancks, D.C.; Elde, N.C.; Cristea, I.M.; Knipe, D.M. CGAS-Mediated Stabilization of IFI16 Promotes Innate Signaling during Herpes Simplex Virus Infection. Proc. Natl. Acad. Sci. USA 2015, 112, E1773–E1781. [Google Scholar] [CrossRef] [Green Version]
- Almine, J.F.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and CGAS Cooperate in the Activation of STING during DNA Sensing in Human Keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef]
- Jønsson, K.L.; Laustsen, A.; Krapp, C.; Skipper, K.A.; Thavachelvam, K.; Hotter, D.; Egedal, J.H.; Kjolby, M.; Mohammadi, P.; Prabakaran, T.; et al. IFI16 Is Required for DNA Sensing in Human Macrophages by Promoting Production and Function of CGAMP. Nat. Commun. 2017, 8, 14391. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Gentili, M.; Lahaye, X.; Nadalin, F.; Nader, G.P.F.; Puig Lombardi, E.; Herve, S.; De Silva, N.S.; Rookhuizen, D.C.; Zueva, E.; Goudot, C.; et al. The N-Terminal Domain of CGAS Determines Preferential Association with Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Rep. 2019, 26, 2377–2393.e13. [Google Scholar] [CrossRef] [Green Version]
- Barnett, K.C.; Coronas-Serna, J.M.; Zhou, W.; Ernandes, M.J.; Cao, A.; Kranzusch, P.J.; Kagan, J.C. Phosphoinositide Interactions Position CGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 2019, 176, 1432–1446.e11. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Huang, T.; Du, M.; Chen, X.; Du, F.; Ren, J.; Chen, Z.J. Phosphorylation and Chromatin Tethering Prevent CGAS Activation during Mitosis. Science 2021, 371, eabc5386. [Google Scholar] [CrossRef]
- Wu, X.; Wu, F.-H.; Wang, X.; Wang, L.; Siedow, J.N.; Zhang, W.; Pei, Z.-M. Molecular Evolutionary and Structural Analysis of the Cytosolic DNA Sensor CGAS and STING. Nucleic Acids Res. 2014, 42, 8243–8257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Lama, L.; Adura, C.; Tomita, D.; Glickman, J.F.; Tuschl, T.; Patel, D.J. Human CGAS Catalytic Domain Has an Additional DNA-Binding Interface That Enhances Enzymatic Activity and Liquid-Phase Condensation. Proc. Natl. Acad. Sci. USA 2019, 116, 11946–11955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranzusch, P.J. CGAS and CD-NTase Enzymes: Structure, Mechanism, and Evolution. Curr. Opin. Struct. Biol. 2019, 59, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Whiteley, A.T.; de Oliveira Mann, C.C.; Morehouse, B.R.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; Mekalanos, J.J.; Kranzusch, P.J. Structure of the Human CGAS–DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 2018, 174, 300–311.e11. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal Roles of CGAS-CGAMP Signaling in Antiviral Defense and Immune Adjuvant Effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef] [Green Version]
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lässig, C.; de Oliveira Mann, C.C.; Jan Drexler, D.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. CGAS Senses Long and HMGB/TFAM-Bound U-Turn DNA by Forming Protein–DNA Ladders. Nature 2017, 549, 394–398. [Google Scholar] [CrossRef]
- Du, M.; Chen, Z.J. DNA-Induced Liquid Phase Condensation of CGAS Activates Innate Immune Signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.-S.; Cai, H.; Xue, W.; Wang, M.; Xia, T.; Li, W.-J.; Xing, J.-Q.; Zhao, M.; Huang, Y.-J.; Chen, S.; et al. G3BP1 Promotes DNA Binding and Activation of CGAS. Nat. Immunol. 2019, 20, 18–28. [Google Scholar] [CrossRef]
- Srikanth, S.; Woo, J.S.; Wu, B.; El-Sherbiny, Y.M.; Leung, J.; Chupradit, K.; Rice, L.; Seo, G.J.; Calmettes, G.; Ramakrishna, C.; et al. The Ca2+ Sensor STIM1 Regulates the Type I Interferon Response by Retaining the Signaling Adaptor STING at the Endoplasmic Reticulum. Nat. Immunol. 2019, 20, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING Is a Direct Innate Immune Sensor of Cyclic Di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef]
- Diner, E.J.; Burdette, D.L.; Wilson, S.C.; Monroe, K.M.; Kellenberger, C.A.; Hyodo, M.; Hayakawa, Y.; Hammond, M.C.; Vance, R.E. The Innate Immune DNA Sensor CGAS Produces a Noncanonical Cyclic Dinucleotide That Activates Human STING. Cell Rep. 2013, 3, 1355–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Dai, T.; He, X.; Zhang, Z.; Xie, F.; Wang, S.; Zhang, L.; Zhou, F. The Interactions between CGAS-STING Pathway and Pathogens. Sig. Transduct. Target. Ther. 2020, 5, 91. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.; Zhang, X. Cryo-EM Structures of STING Reveal Its Mechanism of Activation by Cyclic GMP–AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy Induction via STING Trafficking Is a Primordial Function of the CGAS Pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Stempel, M.; Chan, B.; Juranić Lisnić, V.; Krmpotić, A.; Hartung, J.; Paludan, S.R.; Füllbrunn, N.; Lemmermann, N.A.; Brinkmann, M.M. The Herpesviral Antagonist M152 Reveals Differential Activation of STING -dependent IRF and NF -κB Signaling and STING ’s Dual Role during MCMV Infection. EMBO J. 2019, 38, e100983. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Zou, J.; Saitoh, T.; Kumar, H.; Abe, T.; Matsuura, Y.; Kawai, T.; Akira, S. The Ubiquitin Ligase TRIM56 Regulates Innate Immune Responses to Intracellular Double-Stranded DNA. Immunity 2010, 33, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liu, X.; Cui, Y.; Tang, Y.; Chen, W.; Li, S.; Yu, H.; Pan, Y.; Wang, C. The E3 Ubiquitin Ligase AMFR and INSIG1 Bridge the Activation of TBK1 Kinase by Modifying the Adaptor STING. Immunity 2014, 41, 919–933. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hu, M.-M.; Wang, Y.-Y.; Shu, H.-B. TRIM32 Protein Modulates Type I Interferon Induction and Cellular Antiviral Response by Targeting MITA/STING Protein for K63-Linked Ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Zhang, L.; Shen, J.; Zhai, Y.; Jiang, Q.; Yi, M.; Deng, X.; Ruan, Z.; Fang, R.; Chen, Z.; et al. The STING Phase-Separator Suppresses Innate Immune Signalling. Nat. Cell Biol. 2021, 23, 330–340. [Google Scholar] [CrossRef]
- Agelidis, A.M.; Shukla, D. Cell Entry Mechanisms of HSV: What We Have Learned in Recent Years. Future Virol. 2015, 10, 1145–1154. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Fraser, N.W. Temporal Association of the Herpes Simplex Virus Genome with Histone Proteins during a Lytic Infection. J. Virol. 2008, 82, 3530–3537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorley-Lawson, D.A.; Duca, K.A.; Shapiro, M. Epstein-Barr Virus: A Paradigm for Persistent Infection—For Real and in Virtual Reality. Trends Immunol. 2008, 29, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Weidner-Glunde, M.; Kruminis-Kaszkiel, E.; Savanagouder, M. Herpesviral Latency—Common Themes. Pathogens 2020, 9, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varani, S.; Cederarv, M.; Feld, S.; Tammik, C.; Frascaroli, G.; Landini, M.P.; Söderberg-Nauclér, C. Human Cytomegalovirus Differentially Controls B Cell and T Cell Responses through Effects on Plasmacytoid Dendritic Cells. J. Immunol. 2007, 179, 7767–7776. [Google Scholar] [CrossRef]
- Krug, A.; Luker, G.D.; Barchet, W.; Leib, D.A.; Akira, S.; Colonna, M. Herpes Simplex Virus Type 1 Activates Murine Natural Interferon-Producing Cells through Toll-like Receptor 9. Blood 2004, 103, 1433–1437. [Google Scholar] [CrossRef] [Green Version]
- Zyzak, J.; Mitkiewicz, M.; Leszczyńska, E.; Reniewicz, P.; Moynagh, P.N.; Siednienko, J. HSV-1/TLR9-Mediated IFNβ and TNFα Induction Is Mal-Dependent in Macrophages. J. Innate Immun. 2020, 12, 387–398. [Google Scholar] [CrossRef]
- Hochrein, H.; Schlatter, B.; O’Keeffe, M.; Wagner, C.; Schmitz, F.; Schiemann, M.; Bauer, S.; Suter, M.; Wagner, H. Herpes Simplex Virus Type-1 Induces IFN- Production via Toll-like Receptor 9-Dependent and -Independent Pathways. Proc. Natl. Acad. Sci. USA 2004, 101, 11416–11421. [Google Scholar] [CrossRef] [Green Version]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like Receptor 9–Mediated Recognition of Herpes Simplex Virus-2 by Plasmacytoid Dendritic Cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 Induction of IRF-3 Signaling during Herpesviral Infection and Degradation of IFI16 by the Viral ICP0 Protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Zeng, J.; Fan, S.; Liao, Y.; Feng, M.; Wang, L.; Zhang, Y.; Li, Q. Herpes Simplex Virus Type 1-Encoded MiR-H2-3p Manipulates Cytosolic DNA-Stimulated Antiviral Innate Immune Response by Targeting DDX41. Viruses 2019, 11, 756. [Google Scholar] [CrossRef] [Green Version]
- Justice, J.L.; Kennedy, M.A.; Hutton, J.E.; Liu, D.; Song, B.; Phelan, B.; Cristea, I.M. Systematic Profiling of Protein Complex Dynamics Reveals DNA-PK Phosphorylation of IFI16 En Route to Herpesvirus Immunity. Sci. Adv. 2021, 7, eabg6680. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, L.H.; Wilson, S.C.; Morrison, H.M.; Karalis, V.; Chung, J.-Y.J.; Chen, K.J.; Bateup, H.S.; Szpara, M.L.; Lee, A.Y.; Cox, J.S.; et al. Interferon-Independent STING Signaling Promotes Resistance to HSV-1 in Vivo. Nat. Commun. 2020, 11, 3382. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Dobbs, N.; Yang, K.; Yan, N. Interferon-Independent Activities of Mammalian STING Mediate Antiviral Response and Tumor Immune Evasion. Immunity 2020, 53, 115–126.e5. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 Acts as a Nuclear Pathogen Sensor to Induce the Inflammasome in Response to Kaposi Sarcoma-Associated Herpesvirus Infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.E.; Chikoti, L.; Chandran, B. Herpes Simplex Virus 1 Infection Induces Activation and Subsequent Inhibition of the IFI16 and NLRP3 Inflammasomes. J. Virol. 2013, 87, 5005–5018. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.A.; Singh, V.V.; Dutta, S.; Veettil, M.V.; Dutta, D.; Chikoti, L.; Lu, J.; Everly, D.; Chandran, B. Constitutive Interferon-Inducible Protein 16-Inflammasome Activation during Epstein-Barr Virus Latency I, II, and III in B and Epithelial Cells. J. Virol. 2013, 87, 8606–8623. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, J.; Ansari, M.A.; Kumar, B.; Dutta, D.; Roy, A.; Chikoti, L.; Pisano, G.; Dutta, S.; Vahedi, S.; Veettil, M.V.; et al. Histone H2B-IFI16 Recognition of Nuclear Herpesviral Genome Induces Cytoplasmic Interferon-β Responses. PLoS Pathog. 2016, 12, e1005967. [Google Scholar] [CrossRef] [Green Version]
- Dutta, D.; Dutta, S.; Veettil, M.V.; Roy, A.; Ansari, M.A.; Iqbal, J.; Chikoti, L.; Kumar, B.; Johnson, K.E.; Chandran, B. BRCA1 Regulates IFI16 Mediated Nuclear Innate Sensing of Herpes Viral DNA and Subsequent Induction of the Innate Inflammasome and Interferon-β Responses. PLoS Pathog. 2015, 11, e1005030. [Google Scholar] [CrossRef] [Green Version]
- Su, C.; Zheng, C. Herpes Simplex Virus 1 Abrogates the CGAS/STING-Mediated Cytosolic DNA-Sensing Pathway via Its Virion Host Shutoff Protein, UL41. J. Virol. 2017, 91, e02414-16. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; You, H.; Su, C.; Li, Y.; Chen, S.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates CGAS/STING-Mediated Antiviral Innate Immunity. J. Virol. 2018, 92, e00841-18. [Google Scholar] [CrossRef] [Green Version]
- Teigler, J.E.; Kagan, J.C.; Barouch, D.H. Late Endosomal Trafficking of Alternative Serotype Adenovirus Vaccine Vectors Augments Antiviral Innate Immunity. J. Virol. 2014, 88, 10354–10363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basner-Tschakarjan, E.; Gaffal, E.; O’Keeffe, M.; Tormo, D.; Limmer, A.; Wagner, H.; Hochrein, H.; Tüting, T. Adenovirus Efficiently Transduces Plasmacytoid Dendritic Cells Resulting in TLR9-Dependent Maturation and IFN-α Production. J. Gene Med. 2006, 8, 1300–1306. [Google Scholar] [CrossRef] [PubMed]
- Hensley, S.E.; Giles-Davis, W.; McCoy, K.C.; Weninger, W.; Ertl, H.C.J. Dendritic Cell Maturation, but Not CD8 + T Cell Induction, Is Dependent on Type I IFN Signaling during Vaccination with Adenovirus Vectors. J. Immunol. 2005, 175, 6032–6041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huarte, E.; Larrea, E.; Hernández-Alcoceba, R.; Alfaro, C.; Murillo, O.; Arina, A.; Tirapu, I.; Azpilicueta, A.; Hervás-Stubbs, S.; Bortolanza, S.; et al. Recombinant Adenoviral Vectors Turn on the Type I Interferon System without Inhibition of Transgene Expression and Viral Replication. Mol. Ther. 2006, 14, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, X.; Yang, Y. Innate Immune Response to Adenoviral Vectors Is Mediated by Both Toll-Like Receptor-Dependent and -Independent Pathways. J. Virol. 2007, 81, 3170–3180. [Google Scholar] [CrossRef] [Green Version]
- Iacobelli-Martinez, M.; Nemerow, G.R. Preferential Activation of Toll-Like Receptor Nine by CD46-Utilizing Adenoviruses. J. Virol. 2007, 81, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- McGuire, K.A.; Barlan, A.U.; Griffin, T.M.; Wiethoff, C.M. Adenovirus Type 5 Rupture of Lysosomes Leads to Cathepsin B-Dependent Mitochondrial Stress and Production of Reactive Oxygen Species. J. Virol. 2011, 85, 10806–10813. [Google Scholar] [CrossRef] [Green Version]
- Barlan, A.U.; Griffin, T.M.; Mcguire, K.A.; Wiethoff, C.M. Adenovirus Membrane Penetration Activates the NLRP3 Inflammasome. J. Virol. 2011, 85, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Lam, E.; Stein, S.; Falck-Pedersen, E. Adenovirus Detection by the CGAS/STING/TBK1 DNA Sensing Cascade. J. Virol. 2014, 88, 974–981. [Google Scholar] [CrossRef] [Green Version]
- Stein, S.C.; Falck-Pedersen, E. Sensing Adenovirus Infection: Activation of Interferon Regulatory Factor 3 in RAW 264.7 Cells. J. Virol. 2012, 86, 4527–4537. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Kawabata, K.; Kouyama, E.; Ishii, K.J.; Katayama, K.; Suzuki, T.; Kurachi, S.; Sakurai, F.; Akira, S.; Mizuguchi, H. Induction of Type I Interferon by Adenovirus-Encoded Small RNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 17286–17291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, S.-Y.; Hearing, P. Mechanism of Adenovirus E4-ORF3-Mediated SUMO Modifications. mBio 2019, 10, e00022-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA Tumor Virus Oncogenes Antagonize the CGAS-STING DNA-Sensing Pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Look, D.C.; Roswit, W.T.; Frick, A.G.; Gris-Alevy, Y.; Dickhaus, D.M.; Walter, M.J.; Holtzman, M.J. Direct Suppression of Stat1 Function during Adenoviral Infection. Immunity 1998, 9, 871–880. [Google Scholar] [CrossRef] [Green Version]
- McBride, A.A. Human Papillomaviruses: Diversity, Infection and Host Interactions. Nat. Rev. Microbiol. 2022, 20, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The Biology and Life-Cycle of Human Papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef]
- Day, P.M.; Thompson, C.D.; Schowalter, R.M.; Lowy, D.R.; Schiller, J.T. Identification of a Role for the Trans-Golgi Network in Human Papillomavirus 16 Pseudovirus Infection. J. Virol. 2013, 87, 3862–3870. [Google Scholar] [CrossRef] [Green Version]
- Aydin, I.; Weber, S.; Snijder, B.; Samperio Ventayol, P.; Kühbacher, A.; Becker, M.; Day, P.M.; Schiller, J.T.; Kann, M.; Pelkmans, L.; et al. Large Scale RNAi Reveals the Requirement of Nuclear Envelope Breakdown for Nuclear Import of Human Papillomaviruses. PLoS Pathog. 2014, 10, e1004162. [Google Scholar] [CrossRef]
- Cannella, F.; Pierangeli, A.; Scagnolari, C.; Cacciotti, G.; Tranquilli, G.; Stentella, P.; Recine, N.; Antonelli, G. TLR9 Is Expressed in Human Papillomavirus-Positive Cervical Cells and Is Overexpressed in Persistent Infections. Immunobiology 2015, 220, 363–368. [Google Scholar] [CrossRef]
- Hasan, U.A.; Bates, E.; Takeshita, F.; Biliato, A.; Accardi, R.; Bouvard, V.; Mansour, M.; Vincent, I.; Gissmann, L.; Iftner, T.; et al. TLR9 Expression and Function Is Abolished by the Cervical Cancer-Associated Human Papillomavirus Type 16. J. Immunol. 2007, 178, 3186–3197. [Google Scholar] [CrossRef] [Green Version]
- Reinholz, M.; Kawakami, Y.; Salzer, S.; Kreuter, A.; Dombrowski, Y.; Koglin, S.; Kresse, S.; Ruzicka, T.; Schauber, J. HPV16 Activates the AIM2 Inflammasome in Keratinocytes. Arch. Dermatol. Res. 2013, 305, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Ainouze, M.; Rochefort, P.; Parroche, P.; Roblot, G.; Tout, I.; Briat, F.; Zannetti, C.; Marotel, M.; Goutagny, N.; Auron, P.; et al. Human Papillomavirus Type 16 Antagonizes IRF6 Regulation of IL-1β. PLoS Pathog. 2018, 14, e1007158. [Google Scholar] [CrossRef] [PubMed]
- Uhlorn, B.L.; Jackson, R.; Li, S.; Bratton, S.M.; Van Doorslaer, K.; Campos, S.K. Vesicular Trafficking Permits Evasion of CGAS/STING Surveillance during Initial Human Papillomavirus Infection. PLoS Pathog. 2020, 16, e1009028. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.H.; Bortnik, V.; McMillan, N.A.J.; Idris, A. CGAS-STING Responses Are Dampened in High-Risk HPV Type 16 Positive Head and Neck Squamous Cell Carcinoma Cells. Microb. Pathog. 2019, 132, 162–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, L. Polyomaviruses. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Prado, J.; Monezi, T.; Amorim, A.; Lino, V.; Paladino, A.; Boccardo, E. Human Polyomaviruses and Cancer: An Overview. Clinics 2018, 73. [Google Scholar] [CrossRef]
- Ehlers, B.; Moens, U. Genome Analysis of Non-Human Primate Polyomaviruses. Infect. Genet. Evol. 2014, 26, 283–294. [Google Scholar] [CrossRef]
- Soldatova, I.; Prilepskaja, T.; Abrahamyan, L.; Forstová, J.; Huérfano, S. Interaction of the Mouse Polyomavirus Capsid Proteins with Importins Is Required for Efficient Import of Viral DNA into the Cell Nucleus. Viruses 2018, 10, 165. [Google Scholar] [CrossRef] [Green Version]
- Liebl, D.; Difato, F.; Horníková, L.; Mannová, P.; Štokrová, J.; Forstová, J. Mouse Polyomavirus Enters Early Endosomes, Requires Their Acidic PH for Productive Infection, and Meets Transferrin Cargo in Rab11-Positive Endosomes. J. Virol. 2006, 80, 4610–4622. [Google Scholar] [CrossRef] [Green Version]
- Horníková, L.; Bruštíková, K.; Forstová, J. Microtubules in Polyomavirus Infection. Viruses 2020, 12, 121. [Google Scholar] [CrossRef] [Green Version]
- Huérfano, S.; Ryabchenko, B.; Španielová, H.; Forstová, J. Hydrophobic Domains of Mouse Polyomavirus Minor Capsid Proteins Promote Membrane Association and Virus Exit from the ER. FEBS J. 2017, 284, 883–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padgett, B.L.; Zurhein, G.M.; Walker, D.L.; Eckroade, R.; Dessel, B. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet 1971, 297, 1257–1260. [Google Scholar] [CrossRef]
- Gardner, S.D.; Field, A.M.; Coleman, D.V.; Hulme, B. New human papovavirus (B.K.) Isolated from urine after renal transplantation. Lancet 1971, 297, 1253–1257. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, W.A.; Pipkin, P.; Andrews, N.; Vyse, A.; Minor, P.; Brown, D.W.G.; Miller, E. Population-Based Study of Antibody to the Human Polyomaviruses BKV and JCV and the Simian Polyomavirus SV40. J. Med. Virol. 2003, 71, 115–123. [Google Scholar] [CrossRef]
- Egli, A.; Infanti, L.; Dumoulin, A.; Buser, A.; Samaridis, J.; Stebler, C.; Gosert, R.; Hirsch, H.H. Prevalence of Polyomavirus BK and JC Infection and Replication in 400 Healthy Blood Donors. J. Infect. Dis. 2009, 199, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Šroller, V.; Hamšíková, E.; Ludvíková, V.; Vochozková, P.; Kojzarová, M.; Fraiberk, M.; Saláková, M.; Morávková, A.; Forstová, J.; Němečková, Š. Seroprevalence Rates of BKV, JCV, and MCPyV Polyomaviruses in the General Czech Republic Population: Seroprevalence of BKV, JCV, and MCPyV. J. Med. Virol. 2014, 86, 1560–1568. [Google Scholar] [CrossRef]
- Foulongne, V.; Sauvage, V.; Hebert, C.; Dereure, O.; Cheval, J.; Gouilh, M.A.; Pariente, K.; Segondy, M.; Burguière, A.; Manuguerra, J.-C.; et al. Human Skin Microbiota: High Diversity of DNA Viruses Identified on the Human Skin by High Throughput Sequencing. PLoS ONE 2012, 7, e38499. [Google Scholar] [CrossRef] [Green Version]
- Jouhi, L.; Koljonen, V.; Böhling, T.; Haglund, C.; Hagström, J. The Expression of Toll-like Receptors 2, 4, 5, 7 and 9 in Merkel Cell Carcinoma. Anticancer. Res. 2015, 35, 1843–1849. [Google Scholar]
- Shahzad, N.; Shuda, M.; Gheit, T.; Kwun, H.J.; Cornet, I.; Saidj, D.; Zannetti, C.; Hasan, U.; Chang, Y.; Moore, P.S.; et al. The T Antigen Locus of Merkel Cell Polyomavirus Downregulates Human Toll-Like Receptor 9 Expression. J. Virol. 2013, 87, 13009–13019. [Google Scholar] [CrossRef] [Green Version]
- Ryabchenko, B.; Soldatova, I.; Šroller, V.; Forstová, J.; Huérfano, S. Immune Sensing of Mouse Polyomavirus DNA by P204 and CGAS DNA Sensors. FEBS J. 2021, 288, 5964–5985. [Google Scholar] [CrossRef] [PubMed]
- Krump, N.A.; Wang, R.; Liu, W.; Yang, J.F.; Ma, T.; You, J. Merkel Cell Polyomavirus Infection Induces an Antiviral Innate Immune Response in Human Dermal Fibroblasts. J. Virol. 2021, 95, e02211-20. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yang, R.; Payne, A.S.; Schowalter, R.M.; Spurgeon, M.E.; Lambert, P.F.; Xu, X.; Buck, C.B.; You, J. Identifying the Target Cells and Mechanisms of Merkel Cell Polyomavirus Infection. Cell Host Microbe 2016, 19, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannová, P.; Forstová, J. Mouse Polyomavirus Utilizes Recycling Endosomes for a Traffic Pathway Independent of COPI Vesicle Transport. J. Virol. 2003, 77, 1672–1681. [Google Scholar] [CrossRef] [Green Version]
- de Kort, H.; Heutinck, K.M.; Ruben, J.M.; Ede V. Silva, A.; Wolthers, K.C.; Hamann, J.; ten Berge, I.J.M. Primary Human Renal-Derived Tubular Epithelial Cells Fail to Recognize and Suppress BK Virus Infection. Transplantation 2017, 101, 1820–1829. [Google Scholar] [CrossRef]
- An, P.; Sáenz Robles, M.T.; Duray, A.M.; Cantalupo, P.G.; Pipas, J.M. Human Polyomavirus BKV Infection of Endothelial Cells Results in Interferon Pathway Induction and Persistence. PLoS Pathog. 2019, 15, e1007505. [Google Scholar] [CrossRef]
- Manzetti, J.; Weissbach, F.H.; Graf, F.E.; Unterstab, G.; Wernli, M.; Hopfer, H.; Drachenberg, C.B.; Rinaldo, C.H.; Hirsch, H.H. BK Polyomavirus Evades Innate Immune Sensing by Disrupting the Mitochondrial Network and Promotes Mitophagy. iScience 2020, 23, 101257. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium Taurocholate Cotransporting Polypeptide Is a Functional Receptor for Human Hepatitis B and D Virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Huang, H.-C.; Chen, C.-C.; Chang, W.-C.; Tao, M.-H.; Huang, C. Entry of Hepatitis B Virus into Immortalized Human Primary Hepatocytes by Clathrin-Dependent Endocytosis. J. Virol. 2012, 86, 9443–9453. [Google Scholar] [CrossRef] [Green Version]
- Macovei, A.; Radulescu, C.; Lazar, C.; Petrescu, S.; Durantel, D.; Dwek, R.A.; Zitzmann, N.; Nichita, N.B. Hepatitis B Virus Requires Intact Caveolin-1 Function for Productive Infection in HepaRG Cells. J. Virol. 2010, 84, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Bock, C.T.; Schwinn, S.; Locarnini, S.; Fyfe, J.; Manns, M.P.; Trautwein, C.; Zentgraf, H. Structural Organization of the Hepatitis B Virus Minichromosome. J. Mol. Biol. 2001, 307, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Mutz, P.; Metz, P.; Lempp, F.A.; Bender, S.; Qu, B.; Schöneweis, K.; Seitz, S.; Tu, T.; Restuccia, A.; Frankish, J.; et al. HBV Bypasses the Innate Immune Response and Does Not Protect HCV From Antiviral Activity of Interferon. Gastroenterology 2018, 154, 1791–1804.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suslov, A.; Boldanova, T.; Wang, X.; Wieland, S.; Heim, M.H. Hepatitis B Virus Does Not Interfere With Innate Immune Responses in the Human Liver. Gastroenterology 2018, 154, 1778–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tian, Z. HBV-Induced Immune Imbalance in the Development of HCC. Front. Immunol. 2019, 10, 2048. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, I.; Caux, C.; Hasan, U.; Bendriss-Vermare, N.; Olive, D. Impaired Toll-like Receptor 7 and 9 Signaling: From Chronic Viral Infections to Cancer. Trends Immunol. 2010, 31, 391–397. [Google Scholar] [CrossRef]
- Vincent, I.E.; Zannetti, C.; Lucifora, J.; Norder, H.; Protzer, U.; Hainaut, P.; Zoulim, F.; Tommasino, M.; Trépo, C.; Hasan, U.; et al. Hepatitis B Virus Impairs TLR9 Expression and Function in Plasmacytoid Dendritic Cells. PLoS ONE 2011, 6, e26315. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Hu, Y.; Shi, B.; Zhang, X.; Wang, J.; Zhang, Z.; Shen, F.; Zhang, Q.; Sun, S.; Yuan, Z. HBsAg Inhibits TLR9-Mediated Activation and IFN-α Production in Plasmacytoid Dendritic Cells. Mol. Immunol. 2009, 46, 2640–2646. [Google Scholar] [CrossRef]
- Wu, J.; Lu, M.; Meng, Z.; Trippler, M.; Broering, R.; Szczeponek, A.; Krux, F.; Dittmer, U.; Roggendorf, M.; Gerken, G.; et al. Toll-like Receptor-Mediated Control of HBV Replication by Nonparenchymal Liver Cells in Mice. Hepatology 2007, 46, 1769–1778. [Google Scholar] [CrossRef]
- Martinet, J.; Dufeu–Duchesne, T.; Bruder Costa, J.; Larrat, S.; Marlu, A.; Leroy, V.; Plumas, J.; Aspord, C. Altered Functions of Plasmacytoid Dendritic Cells and Reduced Cytolytic Activity of Natural Killer Cells in Patients With Chronic HBV Infection. Gastroenterology 2012, 143, 1586–1596.e8. [Google Scholar] [CrossRef]
- Verrier, E.R.; Yim, S.; Heydmann, L.; El Saghire, H.; Bach, C.; Turon-Lagot, V.; Mailly, L.; Durand, S.C.; Lucifora, J.; Durantel, D.; et al. Hepatitis B Virus Evasion From Cyclic Guanosine Monophosphate–Adenosine Monophosphate Synthase Sensing in Human Hepatocytes. Hepatology 2018, 68, 1695–1709. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; He, G.; Chen, Y.; Zhang, X. Hepatitis B Virus Might Be Sensed by STING-Dependent DNA Sensors and Attenuates the Response of STING-Dependent DNA Sensing Pathway in Humans with Acute and Chronic Hepatitis B Virus Infection. Viral Immunol. 2020, 33, 642–651. [Google Scholar] [CrossRef]
- Thomsen, M.K.; Nandakumar, R.; Stadler, D.; Malo, A.; Valls, R.M.; Wang, F.; Reinert, L.S.; Dagnaes-Hansen, F.; Hollensen, A.K.; Mikkelsen, J.G.; et al. Lack of Immunological DNA Sensing in Hepatocytes Facilitates Hepatitis B Virus Infection. Hepatology 2016, 64, 746–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauterbach-Rivière, L.; Bergez, M.; Mönch, S.; Qu, B.; Riess, M.; Vondran, F.W.R.; Liese, J.; Hornung, V.; Urban, S.; König, R. Hepatitis B Virus DNA Is a Substrate for the CGAS/STING Pathway but Is Not Sensed in Infected Hepatocytes. Viruses 2020, 12, 592. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, J.; Chen, J.; Li, Y.; Wang, W.; Du, X.; Song, W.; Zhang, W.; Lin, L.; Yuan, Z. Hepatitis B Virus Polymerase Disrupts K63-Linked Ubiquitination of STING To Block Innate Cytosolic DNA-Sensing Pathways. J. Virol. 2015, 89, 2287–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Tang, L.; Cheng, J.; Zhou, T.; Li, Y.; Chang, J.; Zhao, Q.; Guo, J.-T. Hepatitis B Virus Nucleocapsid Uncoating: Biological Consequences and Regulation by Cellular Nucleases. Emerg. Microbes Infect. 2021, 10, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; He, G.; Chen, Y.; Zhang, X.; Wu, S. Differential Activation of NLRP3, AIM2, and IFI16 Inflammasomes in Humans with Acute and Chronic Hepatitis B. Viral Immunol. 2018, 31, 639–645. [Google Scholar] [CrossRef]
- Lu, Y.-Q.; Wu, J.; Wu, X.-J.; Ma, H.; Ma, Y.-X.; Zhang, R.; Su, M.-N.; Wu, N.; Chen, G.-Y.; Chen, H.-S.; et al. Interferon Gamma-Inducible Protein 16 of Peripheral Blood Mononuclear Cells May Sense Hepatitis B Virus Infection and Regulate the Antiviral Immunity. Front. Cell. Infect. Microbiol. 2021, 11, 790036. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, X.; Wang, Z.; Shu, W.; Li, L.; Li, Y.; Guo, Z.; Gao, B.; Xiong, S. Nuclear Sensor Interferon-Inducible Protein 16 Inhibits the Function of Hepatitis B Virus Covalently Closed Circular DNA by Integrating Innate Immune Activation and Epigenetic Suppression. Hepatology 2020, 71, 1154–1169. [Google Scholar] [CrossRef]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell Binding and Entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef]
- Zila, V.; Margiotta, E.; Turoňová, B.; Müller, T.G.; Zimmerli, C.E.; Mattei, S.; Allegretti, M.; Börner, K.; Rada, J.; Müller, B.; et al. Cone-Shaped HIV-1 Capsids Are Transported through Intact Nuclear Pores. Cell 2021, 184, 1032–1046.e18. [Google Scholar] [CrossRef]
- Shen, Q.; Wu, C.; Freniere, C.; Tripler, T.N.; Xiong, Y. Nuclear Import of HIV-1. Viruses 2021, 13, 2242. [Google Scholar] [CrossRef] [PubMed]
- Hatziioannou, T.; Perez-Caballero, D.; Cowan, S.; Bieniasz, P.D. Cyclophilin Interactions with Incoming Human Immunodeficiency Virus Type 1 Capsids with Opposing Effects on Infectivity in Human Cells. J. Virol. 2005, 79, 176–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyamweya, S.; Hegedus, A.; Jaye, A.; Rowland-Jones, S.; Flanagan, K.L.; Macallan, D.C. Comparing HIV-1 and HIV-2 Infection: Lessons for Viral Immunopathogenesis: Comparisons between HIV-1 and HIV-2 Infection. Rev. Med. Virol. 2013, 23, 221–240. [Google Scholar] [CrossRef]
- Lahaye, X.; Manel, N. Viral and Cellular Mechanisms of the Innate Immune Sensing of HIV. Curr. Opin. Virol. 2015, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, G.S.; Raugi, D.N.; Smith, R.A. 90-90-90 for HIV-2? Ending the HIV-2 Epidemic by Enhancing Care and Clinical Management of Patients Infected with HIV-2. Lancet HIV 2018, 5, e390–e399. [Google Scholar] [CrossRef]
- Gao, D.; Wu, J.; Wu, Y.-T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP Synthase Is an Innate Immune Sensor of HIV and Other Retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, M.R.; Bak, R.O.; Andersen, A.; Berg, R.K.; Jensen, S.B.; Jin, T.; Laustsen, A.; Hansen, K.; Ostergaard, L.; Fitzgerald, K.A.; et al. PNAS Plus: From the Cover: IFI16 Senses DNA Forms of the Lentiviral Replication Cycle and Controls HIV-1 Replication. Proc. Natl. Acad. Sci. USA 2013, 110, E4571–E4580. [Google Scholar] [CrossRef] [Green Version]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.T.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 Restriction Factor SAMHD1 Is a Deoxynucleoside Triphosphate Triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef]
- Goujon, C.; Rivière, L.; Jarrosson-Wuilleme, L.; Bernaud, J.; Rigal, D.; Darlix, J.-L.; Cimarelli, A. SIVSM/HIV-2 Vpx Proteins Promote Retroviral Escape from a Proteasome-Dependent Restriction Pathway Present in Human Dendritic Cells. Retrovirology 2007, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx Relieves Inhibition of HIV-1 Infection of Macrophages Mediated by the SAMHD1 Protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [Green Version]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 Is the Dendritic- and Myeloid-Cell-Specific HIV-1 Restriction Factor Counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, X.; Satoh, T.; Gentili, M.; Cerboni, S.; Conrad, C.; Hurbain, I.; El Marjou, A.; Lacabaratz, C.; Lelièvre, J.-D.; Manel, N. The Capsids of HIV-1 and HIV-2 Determine Immune Detection of the Viral CDNA by the Innate Sensor CGAS in Dendritic Cells. Immunity 2013, 39, 1132–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahaye, X.; Gentili, M.; Silvin, A.; Conrad, C.; Picard, L.; Jouve, M.; Zueva, E.; Maurin, M.; Nadalin, F.; Knott, G.J.; et al. NONO Detects the Nuclear HIV Capsid to Promote CGAS-Mediated Innate Immune Activation. Cell 2018, 175, 488–501.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, N.; Regalado-Magdos, A.D.; Stiggelbout, B.; Lee-Kirsch, M.A.; Lieberman, J. The Cytosolic Exonuclease TREX1 Inhibits the Innate Immune Response to Human Immunodeficiency Virus Type 1. Nat. Immunol. 2010, 11, 1005–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Morrison, J.H.; Dingli, D.; Poeschla, E. HIV-1 Activation of Innate Immunity Depends Strongly on the Intracellular Level of TREX1 and Sensing of Incomplete Reverse Transcription Products. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, L.A.; Trifonova, R.T.; Vrbanac, V.; Barteneva, N.S.; Liu, X.; Bollman, B.; Onofrey, L.; Mulik, S.; Ranjbar, S.; Luster, A.D.; et al. TREX1 Knockdown Induces an Interferon Response to HIV That Delays Viral Infection in Humanized Mice. Cell Rep. 2016, 15, 1715–1727. [Google Scholar] [CrossRef] [Green Version]
- Doitsh, G.; Cavrois, M.; Lassen, K.G.; Zepeda, O.; Yang, Z.; Santiago, M.L.; Hebbeler, A.M.; Greene, W.C. Abortive HIV Infection Mediates CD4 T Cell Depletion and Inflammation in Human Lymphoid Tissue. Cell 2010, 143, 789–801. [Google Scholar] [CrossRef] [Green Version]
- Monroe, K.M.; Yang, Z.; Johnson, J.R.; Geng, X.; Doitsh, G.; Krogan, N.J.; Greene, W.C. IFI16 DNA Sensor Is Required for Death of Lymphoid CD4 T Cells Abortively Infected with HIV. Science 2014, 343, 428–432. [Google Scholar] [CrossRef] [Green Version]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jønsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-Canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-ΚB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760.e5. [Google Scholar] [CrossRef] [Green Version]
- Heiser, K.; Nicholas, C.; Garcea, R.L. Activation of DNA Damage Repair Pathways by Murine Polyomavirus. Virology 2016, 497, 346–356. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhao, L.; Gamez, M.; Imperiale, M.J. Roles of ATM and ATR-Mediated DNA Damage Responses during Lytic BK Polyomavirus Infection. PLoS Pathog. 2012, 8, e1002898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowd, G.A.; Li, N.Y.; Fanning, E. ATM and ATR Activities Maintain Replication Fork Integrity during SV40 Chromatin Replication. PLoS Pathog. 2013, 9, e1003283. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sensors | Type of Response | Novel Interacting Partners Identified for the DNA Sensing Pathways | Novel Posttranslational Modifications Identified for the DNA Sensing Pathways |
|---|---|---|---|
| TLR9 | IFN [104,108] | -- | -- |
| IFI16 | IFN | BRCA1, H2B [116,117,118] | IFI16 acetylation [73,118,119] |
| cGAS | IFN | IFI16 [76] | -- |
| IFI16 | Inflammasome | H2B [118] | IFI16 acetylation [118] |
| DDX41 | IFN [15,110] | -- | --- |
| DNA-PK | IFN | IFI16 [20,111] | IFI16 acetylation [20,111] |
| DNA Sensors | Cell Types |
|---|---|
| TLR9 | pDCs [122,123,124,125] Monocytes-TPH-1 [126] Epithelial cells-HeLa [127] |
| cGAS | Macrophages [129] |
| DDX41 | Macrophages [130] |
| RNA Pol III | Fibroblast-MEFs [131] DCs [131] |
| TLR9 | Inflammasome-Related Pathway | STING-Related Pathways | |
|---|---|---|---|
| Mechanism of down regulation | HPV16 E6 and E7 downregulate TLR9 expression [140] | HPV16 E6 and E7 block the IL-1β production [142] |
|
| Pathways | Inflammasome Related Pathways | STING-Related Pathways |
|---|---|---|
| Mechanism of down regulation | HBeAg blocks IL-1β expression [186] |
| Proteins | Function | Influence on the DNA Sensing Response |
|---|---|---|
| SAMHD1 | Depletes cellular pools of deoxynucleotide triphosphates | Down regulate DNA immune sensing by affecting the availability of deoxynucleotides for reverse transcription [198,201] |
| TREX1 | DNA exonuclease | Down regulate immune sensing by degrading HIVDNA intermediates [204,205,206] |
| NONO | Binding to capsids | Promoting sensing by cGAS by unknow mechanism [203] |
| Vpx | Accessory protein | Targets SAMHD1 for degradation [199,200,201] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huérfano, S.; Šroller, V.; Bruštíková, K.; Horníková, L.; Forstová, J. The Interplay between Viruses and Host DNA Sensors. Viruses 2022, 14, 666. https://doi.org/10.3390/v14040666
Huérfano S, Šroller V, Bruštíková K, Horníková L, Forstová J. The Interplay between Viruses and Host DNA Sensors. Viruses. 2022; 14(4):666. https://doi.org/10.3390/v14040666
Chicago/Turabian StyleHuérfano, Sandra, Vojtech Šroller, Kateřina Bruštíková, Lenka Horníková, and Jitka Forstová. 2022. "The Interplay between Viruses and Host DNA Sensors" Viruses 14, no. 4: 666. https://doi.org/10.3390/v14040666