
Transport of Prions in the Peripheral Nervous System: Pathways, Cell Types, and Mechanisms

Abstract
:1. Introduction
2. Prions Spread along Defined Neuroanatomical Pathways
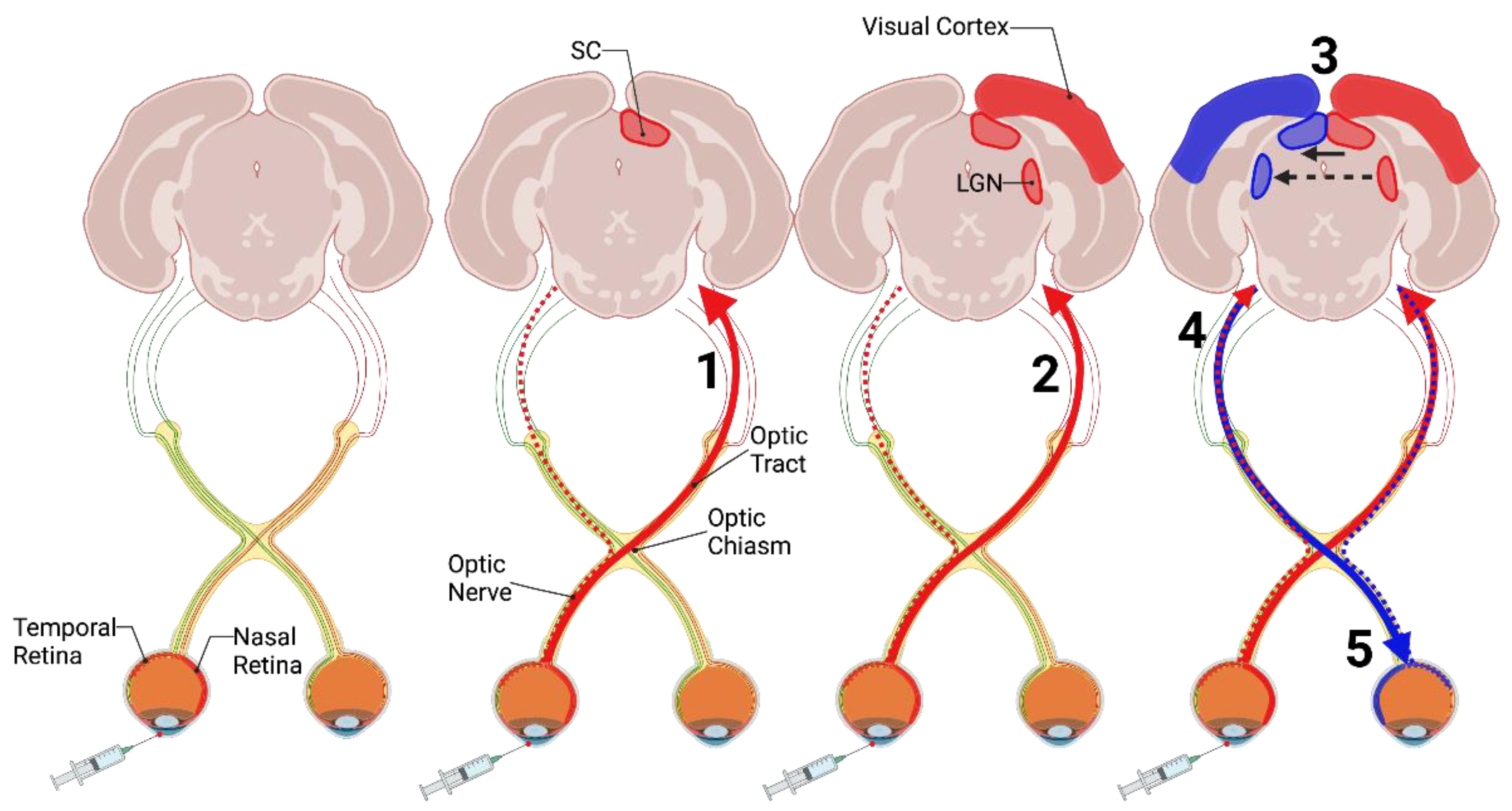
2.1. Intraocular Inoculation of Prions
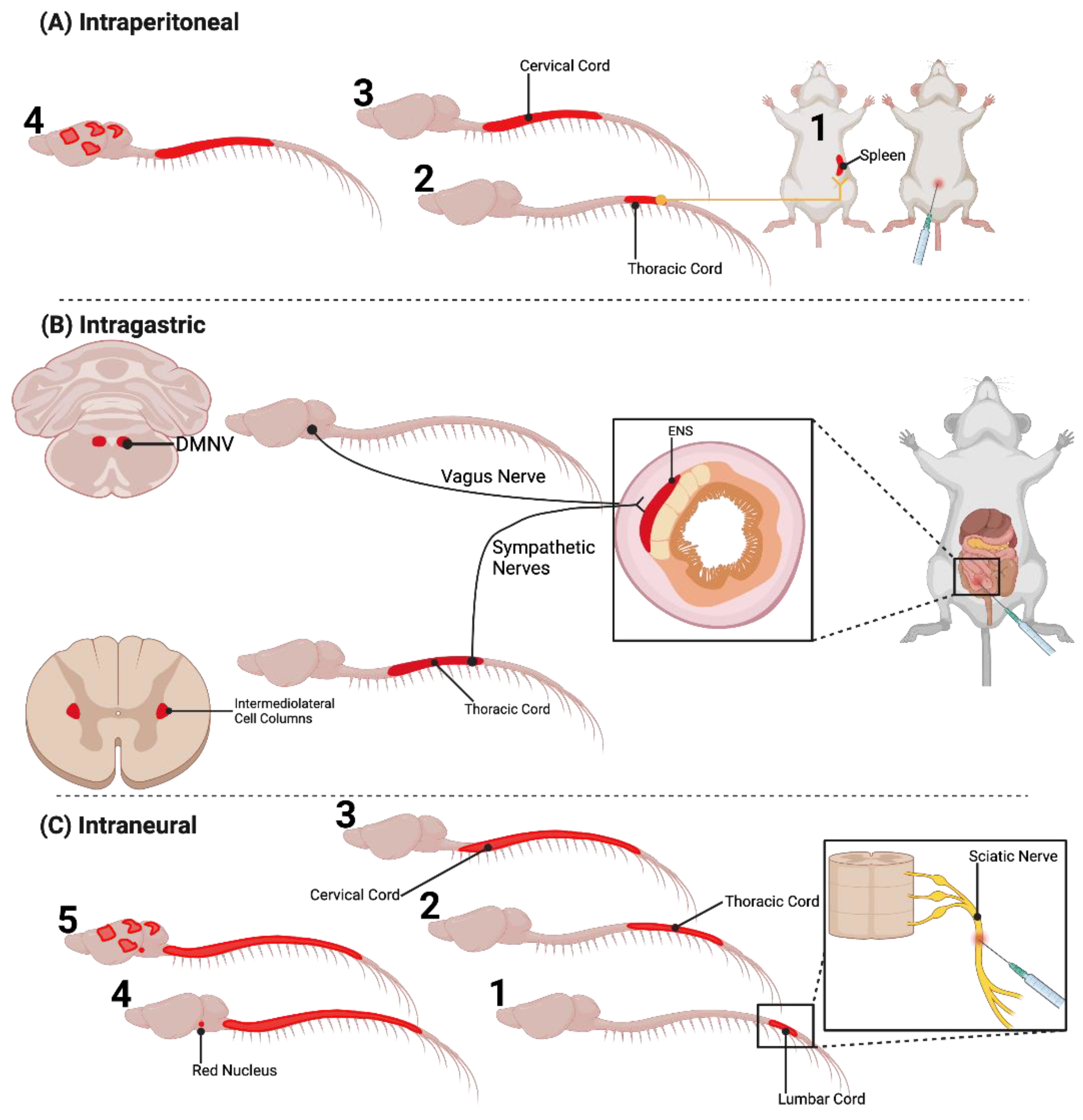
2.2. Extraneural Inoculation of Prions
2.3. Intraneural Inoculation of Prions
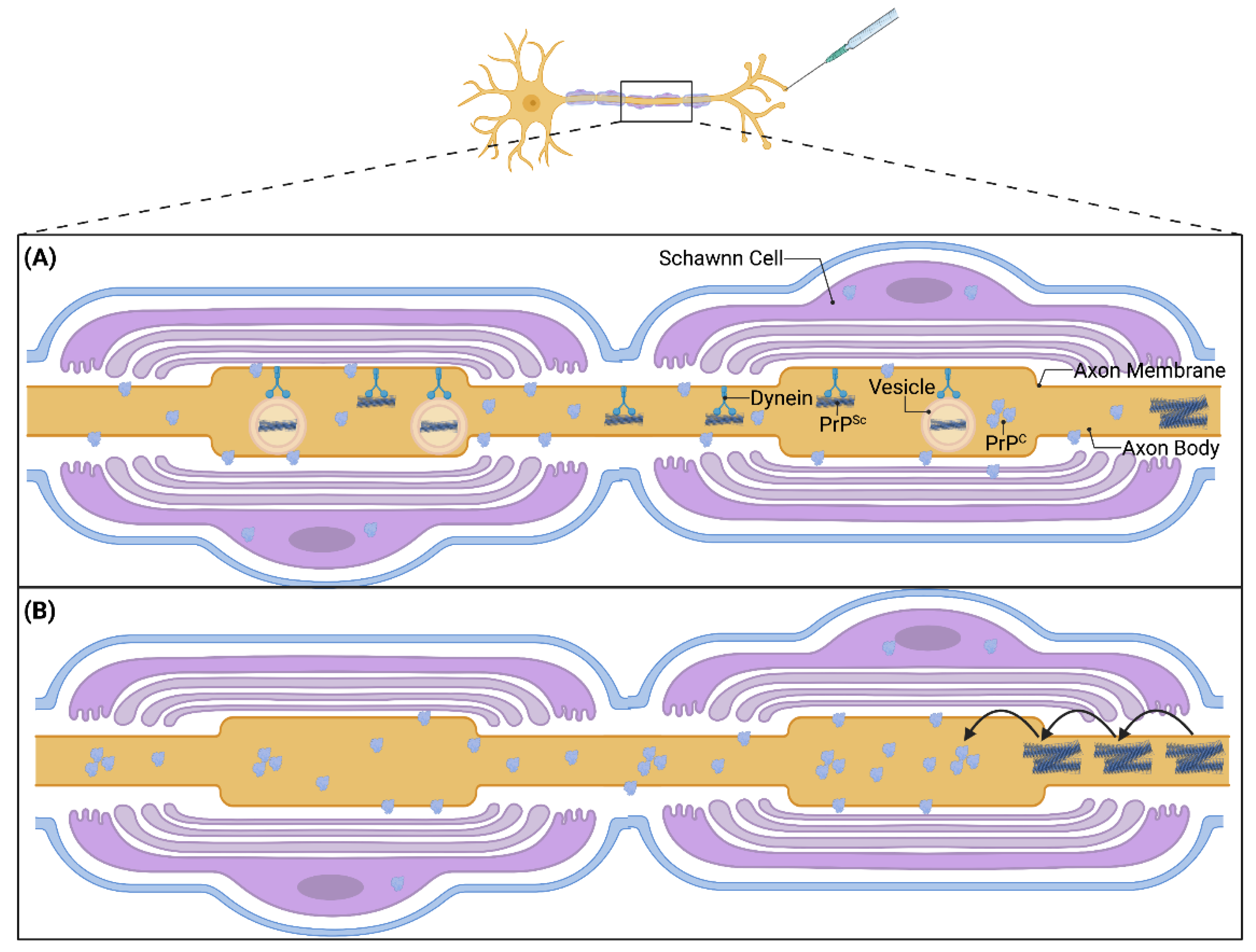
3. Cell Types Involved in Prion Transport
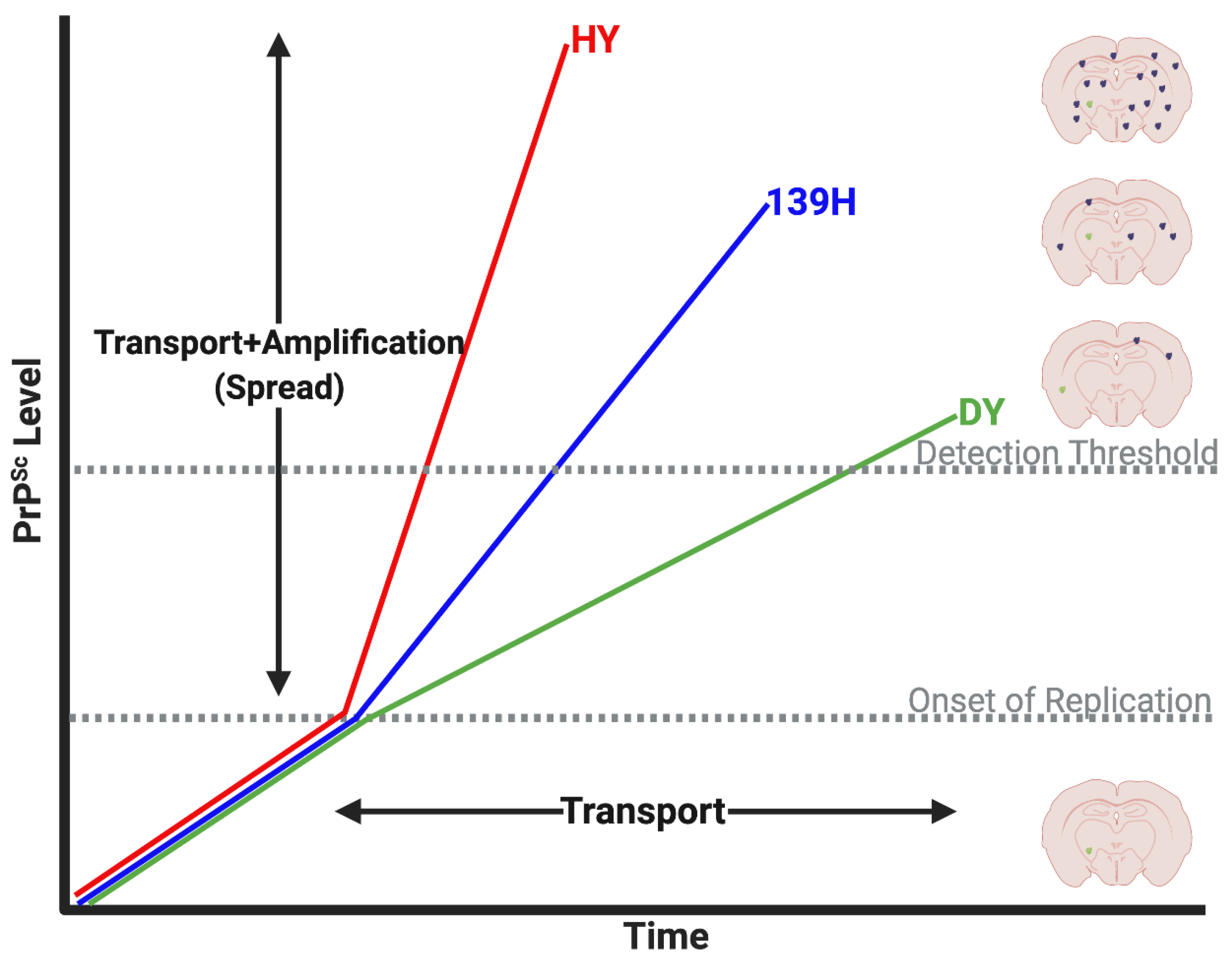
4. Prion Transport Rate and Machinery
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geschwind, M.D. Prion Diseases. Contin. (Minneap. Minn.) 2015, 21, 1612–1638. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, C.J.; Bartz, J.C.; Glatzel, M. Cellular and Molecular Mechanisms of Prion Disease. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 497–516. [Google Scholar] [CrossRef]
- Cole, M. Slow virus diseases. Rocky Mt. Intern. Med. 1975, 72, 294–296. [Google Scholar]
- Fuccillo, D.A.; Kurent, J.E.; Sever, J.L. Slow virus diseases. Annu. Rev. Microbiol. 1974, 28, 231–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajdusek, D.C. Slow virus diseases of the central nervous system. Am. J. Clin. Pathol. 1971, 56, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Hadlow, W.J.; Eklund, C.M. Scrapie—A virus-induced chronic encephalopathy of sheep. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 1968, 44, 281–306. [Google Scholar]
- Ma, J.; Wang, F. Prion disease and the ‘protein-only hypothesis’. Essays Biochem. 2014, 56, 181–191. [Google Scholar] [CrossRef]
- Griffith, J.S. Self-replication and scrapie. Nature 1967, 215, 1043–1044. [Google Scholar] [CrossRef]
- Deleault, N.R.; Harris, B.T.; Rees, J.R.; Supattapone, S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA 2007, 104, 9741–9746. [Google Scholar] [CrossRef] [Green Version]
- Alper, T.; Haig, D.A.; Clarke, M.C. The scrapie agent: Evidence against its dependence for replication on intrinsic nucleic acid. J. Gen. Virol. 1978, 41, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Calzolai, L.; Lysek, D.A.; Pérez, D.R.; Güntert, P.; Wüthrich, K. Prion protein NMR structures of chickens, turtles, and frogs. Proc. Natl. Acad. Sci. USA 2005, 102, 651–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legname, G. Elucidating the function of the prion protein. PLoS Pathog. 2017, 13, e1006458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Funez, P.; Sanchez-Garcia, J.; Rincon-Limas, D.E. Drosophila models of prionopathies: Insight into prion protein function, transmission, and neurotoxicity. Curr. Opin. Genet. Dev. 2017, 44, 141–148. [Google Scholar] [CrossRef]
- Vassallo, N.; Herms, J. Cellular prion protein function in copper homeostasis and redox signalling at the synapse. J. Neurochem. 2003, 86, 538–544. [Google Scholar] [CrossRef]
- Westergard, L.; Christensen, H.M.; Harris, D.A. The cellular prion protein (PrP(C)): Its physiological function and role in disease. Biochim. Biophys. Acta 2007, 1772, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Ironside, J.W.; Ritchie, D.L.; Head, M.W. Prion diseases. Handb. Clin. Neurol. 2017, 145, 393–403. [Google Scholar] [CrossRef]
- Baldwin, K.J.; Correll, C.M. Prion Disease. Semin. Neurol. 2019, 39, 428–439. [Google Scholar] [CrossRef]
- Pattinson, I.H. Resistance of Scrapie Agent to Formalin. J. Comp. Pathol. 1965, 75, 159–164. [Google Scholar] [CrossRef]
- Dickinson, A.G.; Taylor, D.M. Resistance of scrapie agent to decontamination. N. Engl. J. Med. 1978, 299, 1413–1414. [Google Scholar] [CrossRef]
- Gibbs, C.J.; Gajdusek, D.C.; Latarjet, R. Unusual resistance to ionizing radiation of the viruses of kuru, Creutzfeldt-Jakob disease, and scrapie. Proc. Natl. Acad. Sci. USA 1978, 75, 6268–6270. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.; Liberski, P.P.; Wolff, A.; Gajdusek, D.C. Resistance of scrapie infectivity to steam autoclaving after formaldehyde fixation and limited survival after ashing at 360 degrees C: Practical and theoretical implications. J. Infect. Dis. 1990, 161, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Raymond, G.J.; Kocisko, D.A.; Lansbury, P.T. Scrapie infectivity correlates with converting activity, protease resistance, and aggregation of scrapie-associated prion protein in guanidine denaturation studies. J. Virol. 1997, 71, 4107–4110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Q.; Telling, G.; Bartelt-Hunt, S.L.; Bartz, J.C. Dehydration of Prions on Environmentally Relevant Surfaces Protects Them from Inactivation by Freezing and Thawing. J. Virol. 2018, 92, e02191-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, S.E.; Yuan, Q.; Bartz, J.C.; Bartelt-Hunt, S. Effects of solution chemistry and aging time on prion protein adsorption and replication of soil-bound prions. PLoS ONE 2011, 6, e18752. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey, M.; Goodbrand, I.A.; Goodsir, C.M. Pathology of the transmissible spongiform encephalopathies with special emphasis on ultrastructure. Micron 1995, 26, 277–298. [Google Scholar] [CrossRef]
- Gajdusek, D.C.; Zigas, V. Studies on the pathogenesis of kuru; a clinical, pathological and epidemiological study of a chronic, progressive, degenerative disease of the central nervous system achieving epidemic proportions among the natives of the Eastern Highlands of New Guinea. Klin. Wochenschr. 1958, 36, 445–459. [Google Scholar] [CrossRef]
- Morales, R. Prion strains in mammals: Different conformations leading to disease. PLoS Pathog. 2017, 13, e1006323. [Google Scholar] [CrossRef] [Green Version]
- Bartz, J.C. Prion Strain Diversity. Cold Spring Harb. Perspect. Med. 2016, 6, a024349. [Google Scholar] [CrossRef]
- Hill, A.F.; Collinge, J. Prion strains and species barriers. Contrib. Microbiol. 2004, 11, 33–49. [Google Scholar] [CrossRef]
- Aguzzi, A.; Heikenwalder, M.; Polymenidou, M. Insights into prion strains and neurotoxicity. Nat. Rev. Mol. Cell Biol. 2007, 8, 552–561. [Google Scholar] [CrossRef]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissmann, C. Thoughts on mammalian prion strains. Folia Neuropathol. 2009, 47, 104–113. [Google Scholar] [PubMed]
- Shikiya, R.A.; Langenfeld, K.A.; Eckland, T.E.; Trinh, J.; Holec, S.A.M.; Mathiason, C.K.; Kincaid, A.E.; Bartz, J.C. PrPSc formation and clearance as determinants of prion tropism. PLoS Pathog. 2017, 13, e1006298. [Google Scholar] [CrossRef] [PubMed]
- Cortez, L.M.; Nemani, S.K.; Duque Velásquez, C.; Sriraman, A.; Wang, Y.; Wille, H.; McKenzie, D.; Sim, V.L. Asymmetric-flow field-flow fractionation of prions reveals a strain-specific continuum of quaternary structures with protease resistance developing at a hydrodynamic radius of 15 nm. PLoS Pathog. 2021, 17, e1009703. [Google Scholar] [CrossRef] [PubMed]
- Bett, C.; Lawrence, J.; Kurt, T.D.; Orru, C.; Aguilar-Calvo, P.; Kincaid, A.E.; Surewicz, W.K.; Caughey, B.; Wu, C.; Sigurdson, C.J. Enhanced neuroinvasion by smaller, soluble prions. Acta Neuropathol. Commun. 2017, 5, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulland, T.J. Scrapie and Kuru. Can. Vet. J. 1960, 1, 496. [Google Scholar] [PubMed]
- Gajdusek, D.C.; Zigas, V. Degenerative disease of the central nervous system in New Guinea; the endemic occurrence of kuru in the native population. N. Engl. J. Med. 1957, 257, 974–978. [Google Scholar] [CrossRef] [PubMed]
- Gajdusek, D.C.; Zigas, V. Kuru; clinical, pathological and epidemiological study of an acute progressive degenerative disease of the central nervous system among natives of the Eastern Highlands of New Guinea. Am. J. Med. 1959, 26, 442–469. [Google Scholar] [CrossRef]
- Gajdusek, D.C.; Reid, L.H. Studies on kuru. IV. The kuru pattern in Moke, a representative Fore village. Am. J. Trop. Med. Hyg. 1961, 10, 628–638. [Google Scholar] [CrossRef]
- Gajdusek, D.C.; Zigas, V. Studies on kuru. I. The ethnologic setting of kuru. Am. J. Trop. Med. Hyg. 1961, 10, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Gajdusek, D.C.; Zigas, V.; Baker, J. Studies on kuru. III. Patterns of kuru incidence: Demographic and geographic epidemiological analysis. Am. J. Trop. Med. Hyg. 1961, 10, 599–627. [Google Scholar] [CrossRef]
- Gajdusek, D.C. Kuru: An appraisal of five years of investigation. Eugen. Q. 1962, 9, 69–74. [Google Scholar] [CrossRef]
- Gajdusek, D.C. Kuru. Trans. R. Soc. Trop. Med. Hyg. 1963, 57, 151–169. [Google Scholar] [CrossRef]
- Liberski, P.P.; Gajos, A.; Sikorska, B.; Lindenbaum, S. Kuru, the First Human Prion Disease. Viruses 2019, 11, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, G.A.; Scott, A.C.; Johnson, C.T.; Gunning, R.F.; Hancock, R.D.; Jeffrey, M.; Dawson, M.; Bradley, R. A novel progressive spongiform encephalopathy in cattle. Vet. Rec. 1987, 121, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Wilesmith, J.W.; Wells, G.A.; Cranwell, M.P.; Ryan, J.B. Bovine spongiform encephalopathy: Epidemiological studies. Vet. Rec. 1988, 123, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Wilesmith, J.W.; Ryan, J.B.; Atkinson, M.J. Bovine spongiform encephalopathy: Epidemiological studies on the origin. Vet. Rec. 1991, 128, 199–203. [Google Scholar] [CrossRef]
- Bruce, M.E.; Will, R.G.; Ironside, J.W.; McConnell, I.; Drummond, D.; Suttie, A.; McCardle, L.; Chree, A.; Hope, J.; Birkett, C.; et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 1997, 389, 498–501. [Google Scholar] [CrossRef]
- Hill, A.F.; Desbruslais, M.; Joiner, S.; Sidle, K.C.; Gowland, I.; Collinge, J.; Doey, L.J.; Lantos, P. The same prion strain causes vCJD and BSE. Nature 1997, 389, 448–450. [Google Scholar] [CrossRef]
- Houston, F.; Andréoletti, O. Animal prion diseases: The risks to human health. Brain Pathol. 2019, 29, 248–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartsough, G.R.; Burger, D. Encephalopathy of mink. I. Epizootiologic and clinical observations. J. Infect. Dis. 1965, 115, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Hanson, R.P.; Eckroade, R.J.; Marsh, R.F.; Zu Rhein, G.M.; Kanitz, C.L.; Gustafson, D.P. Susceptibility of mink to sheep scrapie. Science 1971, 172, 859–861. [Google Scholar] [CrossRef] [PubMed]
- Marsh, R.F.; Hanson, R.P. On the origin of transmissible mink encephalopathy. In Slow Transmissible Diseases of the Nervous System; Prusiner, S.B., Hadlow, W.J., Eds.; Academic Press: New York, NY, USA, 1979; Volume 1, pp. 451–460. [Google Scholar]
- Marsh, R.F.; Bessen, R.A.; Lehmann, S.; Hartsough, G.R. Epidemiological and experimental studies on a new incident of transmissible mink encephalopathy. J. Gen. Virol. 1991, 72 Pt 3, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Marsh, R.F.; Bessen, R.A. Epidemiologic and experimental studies on transmissible mink encephalopathy. Dev. Biol. Stand. 1993, 80, 111–118. [Google Scholar] [PubMed]
- Baron, T.; Bencsik, A.; Biacabe, A.G.; Morignat, E.; Bessen, R.A. Phenotypic similarity of transmissible mink encephalopathy in cattle and L-type bovine spongiform encephalopathy in a mouse model. Emerg. Infect. Dis. 2007, 13, 1887–1894. [Google Scholar] [CrossRef]
- Adcock, J.L.; Keiss, R.E. Locoism in elk. A disease resembling cerebral pseudolipidosis. Bull. Wildl. Dis. Assoc. 1969, 5, 121–124. [Google Scholar] [CrossRef]
- Williams, E.S.; Young, S. Chronic wasting disease of captive mule deer: A spongiform encephalopathy. J. Wildl. Dis. 1980, 16, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Sigurdson, C.J.; Aguzzi, A. Chronic wasting disease. Biochim. Biophys. Acta 2007, 1772, 610–618. [Google Scholar] [CrossRef]
- CDC. Chronic Wasting Disease (CWD), Occurrence, January 2022. Available online: https://www.cdc.gov/prions/cwd/occurrence.html (accessed on 23 February 2022).
- Benestad, S.L.; Mitchell, G.; Simmons, M.; Ytrehus, B.; Vikøren, T. First case of chronic wasting disease in Europe in a Norwegian free-ranging reindeer. Vet. Res. 2016, 47, 88. [Google Scholar] [CrossRef]
- Pirisinu, L.; Tran, L.; Chiappini, B.; Vanni, I.; Di Bari, M.A.; Vaccari, G.; Vikøren, T.; Madslien, K.I.; Våge, J.; Spraker, T.; et al. Novel Type of Chronic Wasting Disease Detected in Moose (Alces alces), Norway. Emerg. Infect. Dis. 2018, 24, 2210–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonno, R.; Di Bari, M.A.; Pirisinu, L.; D’Agostino, C.; Vanni, I.; Chiappini, B.; Marcon, S.; Riccardi, G.; Tran, L.; Vikøren, T.; et al. Studies in bank voles reveal strain differences between chronic wasting disease prions from Norway and North America. Proc. Natl. Acad. Sci. USA 2020, 117, 31417–31426. [Google Scholar] [CrossRef] [PubMed]
- Ågren, E.O.; Sörén, K.; Gavier-Widén, D.; Benestad, S.L.; Tran, L.; Wall, K.; Averhed, G.; Doose, N.; Våge, J.; Nöremark, M. First Detection of Chronic Wasting Disease in Moose (Alces alces) in Sweden. J. Wildl. Dis. 2021, 57, 461–463. [Google Scholar] [CrossRef]
- Güere, M.E.; Våge, J.; Tharaldsen, H.; Kvie, K.S.; Bårdsen, B.J.; Benestad, S.L.; Vikøren, T.; Madslien, K.; Rolandsen, C.M.; Tranulis, M.A.; et al. Chronic wasting disease in Norway-A survey of prion protein gene variation among cervids. Transbound. Emerg. Dis. 2021, 1–12. [Google Scholar] [CrossRef]
- Tranulis, M.A.; Gavier-Widén, D.; Våge, J.; Nöremark, M.; Korpenfelt, S.L.; Hautaniemi, M.; Pirisinu, L.; Nonno, R.; Benestad, S.L. Chronic wasting disease in Europe: New strains on the horizon. Acta Vet. Scand. 2021, 63, 48. [Google Scholar] [CrossRef]
- Williams, E.S. Chronic wasting disease. Vet. Pathol. 2005, 42, 530–549. [Google Scholar] [CrossRef]
- Detwiler, L.A. Scrapie. Rev. Sci. Tech. 1992, 11, 491–537. [Google Scholar] [CrossRef] [Green Version]
- Plummer, P.J. Scrapie-A Disease of Sheep: A Review of the literature. Can. J. Comp. Med. Vet Sci. 1946, 10, 49–54. [Google Scholar]
- Dickinson, A.G.; Mackay, J.M. Genetical Control of the Incubation Period in Mice of the Neurological Disease, Scrapie. Heredity 1964, 19, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickinson, A.G.; Mackay, J.M.; Zlotnik, I. Transmission by Contact of Scrapie in Mice. J. Comp. Pathol. 1964, 74, 250–254. [Google Scholar] [CrossRef]
- Dickinson, A.G.; Young, G.B.; Stamp, J.T.; Renwick, C.C. An analysis of natural scrapie in Suffolk sheep. Heredity 1965, 20, 485–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickinson, A.G.; Meikle, V.M.; Fraser, H. Identification of a gene which controls the incubation period of some strains of scrapie agent in mice. J. Comp. Pathol. 1968, 78, 293–299. [Google Scholar] [CrossRef]
- Dickinson, A.G.; Stamp, J.T.; Renwick, C.C.; Rennie, J.C. Some factors controlling the incidence of scrapie in Cheviot sheep injected with a Cheviot-passaged scrapie agent. J. Comp. Pathol. 1968, 78, 313–321. [Google Scholar] [CrossRef]
- Kimberlin, R.H. Biochemical changes in scrapie affeced brain. Biochem. J. 1969, 114, 20P–22P. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgsson, G.; Sigurdarson, S.; Brown, P. Infectious agent of sheep scrapie may persist in the environment for at least 16 years. J. Gen. Virol. 2006, 87, 3737–3740. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.E.; Bartelt-Hunt, S.L.; Bartz, J.C. Prions in the environment: Occurrence, fate and mitigation. Prion 2008, 2, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, S.A.; Simmons, H.A.; Gough, K.C.; Maddison, B.C. Persistence of ovine scrapie infectivity in a farm environment following cleaning and decontamination. Vet. Rec. 2015, 176, 99. [Google Scholar] [CrossRef]
- Andréoletti, O.; Lacroux, C.; Chabert, A.; Monnereau, L.; Tabouret, G.; Lantier, F.; Berthon, P.; Eychenne, F.; Lafond-Benestad, S.; Elsen, J.M.; et al. PrP(Sc) accumulation in placentas of ewes exposed to natural scrapie: Influence of foetal PrP genotype and effect on ewe-to-lamb transmission. J. Gen. Virol. 2002, 83, 2607–2616. [Google Scholar] [CrossRef]
- Buyukmihci, N.; Goehring-Harmon, F.; Marsh, R.F. Neural pathogenesis of experimental scrapie after intraocular inoculation of hamsters. Exp. Neurol. 1983, 81, 396–406. [Google Scholar] [CrossRef]
- Fraser, H.; Dickinson, A.G. Targeting of scrapie lesions and spread of agent via the retino-tectal projection. Brain Res. 1985, 346, 32–41. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Walker, C.A. Pathogenesis of scrapie (strain 263K) in hamsters infected intracerebrally, intraperitoneally or intraocularly. J. Gen. Virol. 1986, 67 Pt 2, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.R.; Davies, D.; Fraser, H. Scrapie in the central nervous system: Neuroanatomical spread of infection and Sinc control of pathogenesis. J. Gen. Virol. 1992, 73 Pt 7, 1637–1644. [Google Scholar] [CrossRef]
- Scott, J.R.; Fraser, H. Transport and targeting of scrapie infectivity and pathology in the optic nerve projections following intraocular infection. Prog. Clin. Biol. Res. 1989, 317, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Sefton, A.J.; Dreher, B. Visual System. In The Rat Nervous System, Forebrain and Midbrain; Paxinos, G., Ed.; Academic Press Inc.: Orlando, FL, USA, 1985; Volume 1, pp. 169–210. [Google Scholar]
- Jeffery, G. Retinal ganglion cell death and terminal field retraction in the developing rodent visual system. Brain Res. 1984, 315, 81–96. [Google Scholar] [CrossRef]
- Dreher, B.; Sefton, A.J.; Ni, S.Y.; Nisbett, G. The morphology, number, distribution and central projections of Class I retinal ganglion cells in albino and hooded rats. Brain Behav. Evol. 1985, 26, 10–48. [Google Scholar] [CrossRef] [PubMed]
- Fish, S.E.; Goodman, D.K.; Kuo, D.C.; Polcer, J.D.; Rhoades, R.W. The intercollicular pathway in the golden hamster: An anatomical study. J. Comp. Neurol. 1982, 204, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Mackay-Sim, A.; Sefton, A.J.; Martin, P.R. Subcortical projections to lateral geniculate and thalamic reticular nuclei in the hooded rat. J. Comp. Neurol. 1983, 213, 24–35. [Google Scholar] [CrossRef]
- Hubel, D.H.; Wiesel, T.N. Cortical and callosal connections concerned with the vertical meridian of visual fields in the cat. J. Neurophysiol. 1967, 30, 1561–1573. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Walker, C.A. Pathogenesis of mouse scrapie: Dynamics of agent replication in spleen, spinal cord and brain after infection by different routes. J. Comp. Pathol. 1979, 89, 551–562. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Walker, C.A. Pathogenesis of mouse scrapie: Evidence for neural spread of infection to the CNS. J. Gen. Virol. 1980, 51, 183–187. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Walker, C.A. The role of the spleen in the neuroinvasion of scrapie in mice. Virus Res. 1989, 12, 201–211. [Google Scholar] [CrossRef]
- Muramoto, T.; Kitamoto, T.; Tateishi, J.; Goto, I. The sequential development of abnormal prion protein accumulation in mice with Creutzfeldt-Jakob disease. Am. J. Pathol. 1992, 140, 1411–1420. [Google Scholar]
- Brown, K.L.; Stewart, K.; Ritchie, D.L.; Mabbott, N.A.; Williams, A.; Fraser, H.; Morrison, W.I.; Bruce, M.E. Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat. Med. 1999, 5, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Stewart, K.; Ritchie, D.; Fraser, H.; Morrison, W.I.; Bruce, M.E. Follicular dendritic cells in scrapie pathogenesis. Arch. Virol. Suppl. 2000, 16, 13–21. [Google Scholar] [CrossRef]
- Bruce, M.E.; Brown, K.L.; Mabbott, N.A.; Farquhar, C.F.; Jeffrey, M. Follicular dendritic cells in TSE pathogenesis. Immunol. Today 2000, 21, 442–446. [Google Scholar] [CrossRef]
- Fraser, H.; Dickinson, A.G. Studies of the lymphoreticular system in the pathogenesis of scrapie: The role of spleen and thymus. J. Comp. Pathol. 1978, 88, 563–573. [Google Scholar] [CrossRef]
- Aguzzi, A.; Nuvolone, M.; Zhu, C. The immunobiology of prion diseases. Nat. Rev. Immunol. 2013, 13, 888–902. [Google Scholar] [CrossRef]
- McCulloch, L.; Brown, K.L.; Bradford, B.M.; Hopkins, J.; Bailey, M.; Rajewsky, K.; Manson, J.C.; Mabbott, N.A. Follicular dendritic cell-specific prion protein (PrP) expression alone is sufficient to sustain prion infection in the spleen. PLoS Pathog. 2011, 7, e1002402. [Google Scholar] [CrossRef] [Green Version]
- Castro-Seoane, R.; Hummerich, H.; Sweeting, T.; Tattum, M.H.; Linehan, J.M.; Fernandez de Marco, M.; Brandner, S.; Collinge, J.; Klöhn, P.C. Plasmacytoid dendritic cells sequester high prion titres at early stages of prion infection. PLoS Pathog. 2012, 8, e1002538. [Google Scholar] [CrossRef] [Green Version]
- Prinz, M.; Heikenwalder, M.; Junt, T.; Schwarz, P.; Glatzel, M.; Heppner, F.L.; Fu, Y.X.; Lipp, M.; Aguzzi, A. Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 2003, 425, 957–962. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Walker, C.A. Pathogenesis of mouse scrapie: Patterns of agent replication in different parts of the CNS following intraperitoneal infection. J. R. Soc. Med. 1982, 75, 618–624. [Google Scholar] [PubMed]
- Kimberlin, R.H.; Walker, C.A. Pathogenesis of scrapie in mice after intragastric infection. Virus Res. 1989, 12, 213–220. [Google Scholar] [CrossRef]
- van Keulen, L.J.; Schreuder, B.E.; Vromans, M.E.; Langeveld, J.P.; Smits, M.A. Pathogenesis of natural scrapie in sheep. Arch. Virol. Suppl. 2000, 16, 57–71. [Google Scholar] [CrossRef]
- Kaatz, M.; Fast, C.; Ziegler, U.; Balkema-Buschmann, A.; Hammerschmidt, B.; Keller, M.; Oelschlegel, A.; McIntyre, L.; Groschup, M.H. Spread of classic BSE prions from the gut via the peripheral nervous system to the brain. Am. J. Pathol. 2012, 181, 515–524. [Google Scholar] [CrossRef]
- Ackermann, I.; Ulrich, R.; Tauscher, K.; Fatola, O.I.; Keller, M.; Shawulu, J.C.; Arnold, M.; Czub, S.; Groschup, M.H.; Balkema-Buschmann, A. Prion Infectivity and PrP. Int. J. Mol. Sci. 2021, 22, 1310. [Google Scholar] [CrossRef]
- Hoffmann, C.; Ziegler, U.; Buschmann, A.; Weber, A.; Kupfer, L.; Oelschlegel, A.; Hammerschmidt, B.; Groschup, M.H. Prions spread via the autonomic nervous system from the gut to the central nervous system in cattle incubating bovine spongiform encephalopathy. J. Gen. Virol. 2007, 88, 1048–1055. [Google Scholar] [CrossRef]
- Beekes, M.; McBride, P.A. Early accumulation of pathological PrP in the enteric nervous system and gut-associated lymphoid tissue of hamsters orally infected with scrapie. Neurosci. Lett. 2000, 278, 181–184. [Google Scholar] [CrossRef]
- McBride, P.A.; Beekes, M. Pathological PrP is abundant in sympathetic and sensory ganglia of hamsters fed with scrapie. Neurosci. Lett. 1999, 265, 135–138. [Google Scholar] [CrossRef]
- Beekes, M.; Baldauf, E.; Diringer, H. Sequential appearance and accumulation of pathognomonic markers in the central nervous system of hamsters orally infected with scrapie. J. Gen. Virol. 1996, 77 Pt 8, 1925–1934. [Google Scholar] [CrossRef]
- Beekes, M.; McBride, P.A.; Baldauf, E. Cerebral targeting indicates vagal spread of infection in hamsters fed with scrapie. J. Gen. Virol. 1998, 79 Pt 3, 601–607. [Google Scholar] [CrossRef]
- McBride, P.A.; Schulz-Schaeffer, W.J.; Donaldson, M.; Bruce, M.; Diringer, H.; Kretzschmar, H.A.; Beekes, M. Early spread of scrapie from the gastrointestinal tract to the central nervous system involves autonomic fibers of the splanchnic and vagus nerves. J. Virol. 2001, 75, 9320–9327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, K.A.; Jewell, J.E.; Williams, E.S.; Miller, M.W. Patterns of PrPCWD accumulation during the course of chronic wasting disease infection in orally inoculated mule deer (Odocoileus hemionus). J. Gen. Virol. 2006, 87, 3451–3461. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, C.J.; Spraker, T.R.; Miller, M.W.; Oesch, B.; Hoover, E.A. PrP(CWD) in the myenteric plexus, vagosympathetic trunk and endocrine glands of deer with chronic wasting disease. J. Gen. Virol. 2001, 82, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Hall, S.M.; Walker, C.A. Pathogenesis of mouse scrapie. Evidence for direct neural spread of infection to the CNS after injection of sciatic nerve. J. Neurol. Sci. 1983, 61, 315–325. [Google Scholar] [CrossRef]
- Bartz, J.C.; Kincaid, A.E.; Bessen, R.A. Retrograde transport of transmissible mink encephalopathy within descending motor tracts. J. Virol. 2002, 76, 5759–5768. [Google Scholar] [CrossRef] [Green Version]
- Ayers, J.I.; Kincaid, A.E.; Bartz, J.C. Prion strain targeting independent of strain-specific neuronal tropism. J. Virol. 2009, 83, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Kratzel, C.; Mai, J.; Madela, K.; Beekes, M.; Krüger, D. Propagation of scrapie in peripheral nerves after footpad infection in normal and neurotoxin exposed hamsters. Vet. Res. 2007, 38, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langenfeld, K.A.; Shikiya, R.A.; Kincaid, A.E.; Bartz, J.C. Incongruity between Prion Conversion and Incubation Period following Coinfection. J. Virol. 2016, 90, 5715–5723. [Google Scholar] [CrossRef] [Green Version]
- Bartz, J.C.; Kincaid, A.E.; Bessen, R.A. Rapid prion neuroinvasion following tongue infection. J. Virol. 2003, 77, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Davis, B.M.; Rall, G.F.; Schnell, M.J. Everything You Always Wanted to Know About Rabies Virus (But Were Afraid to Ask). Annu. Rev. Virol. 2015, 2, 451–471. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Miranda-Saksena, M.; Douglas, M.W.; Cunningham, A.L. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 2008, 18, 35–51. [Google Scholar] [CrossRef]
- Follet, J.; Lemaire-Vieille, C.; Blanquet-Grossard, F.; Podevin-Dimster, V.; Lehmann, S.; Chauvin, J.P.; Decavel, J.P.; Varea, R.; Grassi, J.; Fontès, M.; et al. PrP expression and replication by Schwann cells: Implications in prion spreading. J. Virol. 2002, 76, 2434–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groschup, M.H.; Beekes, M.; McBride, P.A.; Hardt, M.; Hainfellner, J.A.; Budka, H. Deposition of disease-associated prion protein involves the peripheral nervous system in experimental scrapie. Acta Neuropathol. 1999, 98, 453–457. [Google Scholar] [CrossRef]
- Hainfellner, J.A.; Budka, H. Disease associated prion protein may deposit in the peripheral nervous system in human transmissible spongiform encephalopathies. Acta Neuropathol. 1999, 98, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, A.C.; Baron, G.S.; Lee, K.S.; Steele-Mortimer, O.; Dorward, D.; Prado, M.A.; Caughey, B. Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J. Neurosci. 2005, 25, 5207–5216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, B.M.; Tuzi, N.L.; Feltri, M.L.; McCorquodale, C.; Cancellotti, E.; Manson, J.C. Dramatic reduction of PrP C level and glycosylation in peripheral nerves following PrP knock-out from Schwann cells does not prevent transmissible spongiform encephalopathy neuroinvasion. J. Neurosci. 2009, 29, 15445–15454. [Google Scholar] [CrossRef] [Green Version]
- Halliez, S.; Chesnais, N.; Mallucci, G.; Vilotte, M.; Langevin, C.; Jaumain, E.; Laude, H.; Vilotte, J.L.; Béringue, V. Targeted knock-down of cellular prion protein expression in myelinating Schwann cells does not alter mouse prion pathogenesis. J. Gen. Virol. 2013, 94, 1435–1440. [Google Scholar] [CrossRef] [Green Version]
- Kovács, G.G.; Preusser, M.; Strohschneider, M.; Budka, H. Subcellular localization of disease-associated prion protein in the human brain. Am. J. Pathol. 2005, 166, 287–294. [Google Scholar] [CrossRef] [Green Version]
- Shearin, H.; Bessen, R.A. Axonal and transynaptic spread of prions. J. Virol. 2014, 88, 8640–8655. [Google Scholar] [CrossRef] [Green Version]
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L. Axonal transport: Cargo-specific mechanisms of motility and regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, L.S.; Yang, Z. Microtubule-based transport systems in neurons: The roles of kinesins and dyneins. Annu. Rev. Neurosci. 2000, 23, 39–71. [Google Scholar] [CrossRef]
- Büeler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Groth, D.; Serban, A.; Koehler, R.; Foster, D.; Torchia, M.; Burton, D.; Yang, S.L.; DeArmond, S.J. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc. Natl. Acad. Sci. USA 1993, 90, 10608–10612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandner, S.; Raeber, A.; Sailer, A.; Blättler, T.; Fischer, M.; Weissmann, C.; Aguzzi, A. Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc. Natl. Acad. Sci. USA 1996, 93, 13148–13151. [Google Scholar] [CrossRef] [Green Version]
- Brandner, S.; Isenmann, S.; Raeber, A.; Fischer, M.; Sailer, A.; Kobayashi, Y.; Marino, S.; Weissmann, C.; Aguzzi, A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996, 379, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Priola, S.A. Cell biology of prion infection. Handb. Clin. Neurol. 2018, 153, 45–68. [Google Scholar] [CrossRef]
- Greil, C.S.; Vorberg, I.M.; Ward, A.E.; Meade-White, K.D.; Harris, D.A.; Priola, S.A. Acute cellular uptake of abnormal prion protein is cell type and scrapie-strain independent. Virology 2008, 379, 284–293. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inoculation Site | Studies | Route of Invasion | Neural Pathways | Targets | Transport | Rate |
|---|---|---|---|---|---|---|
| Intraocular | Buyukmihci et al., 1983 Fraser & Dickinson, 1985 Kimberlin & Walker, 1986 Scott & Fraser, 1989 Scott et al., 1992 | Retinal Ganglion Cells | Optic Nerve Optic tract | SC LGN Visual Cortex | Anterograde | Slow 1.0 mm/day |
| Intraperitoneal | Kimberlin & Walker, 1979 Kimberlin & Walker, 1980 Kimberlin & Walker, 1982 Kimberlin & Walker, 1986 Kimberlin & Walker, 1989b | Spleen | ANS: Splenic Sympathetic Nerves | Brainstem | Retrograde | Slow 0.5–1.0 mm/day |
| Oral/Intragastric | Kimberlin & Walker, 1989a Beekes et al., 1996 Beekes et al., 1998 McBride & Beekes, 1999 Beekes & McBride, 2000 McBride et al., 2001 van Keulen et al., 2000 Sigurdson et al., 2000 Fox et al., 2006 | Peyer’s Patches | ENS: Sympathetic and Parasympathetic (Vagus) Nerves | DMNV Brainstem | Retrograde | Slow 0.8–2.0 mm/day |
| Intraneural | Kimberlin et al., 1983 Bartz et al., 2002 Kratzel et al., 2007 Ayers et al., 2009 Langenfeld et al., 2016 | Sciatic Nerve | Sciatic Nerve Lumbar Spinal Nerves | Lumbar VMNs RN Motor Cortex LVN RF | Retrograde | Slow 1.0–4.0 mm/day |
| Intralingual | Bartz et al., 2003 | Hypoglossal Nerve | Hypoglossal Nerve | Hypoglossal Nucleus | Retrograde | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koshy, S.M.; Kincaid, A.E.; Bartz, J.C. Transport of Prions in the Peripheral Nervous System: Pathways, Cell Types, and Mechanisms. Viruses 2022, 14, 630. https://doi.org/10.3390/v14030630
Koshy SM, Kincaid AE, Bartz JC. Transport of Prions in the Peripheral Nervous System: Pathways, Cell Types, and Mechanisms. Viruses. 2022; 14(3):630. https://doi.org/10.3390/v14030630
Chicago/Turabian StyleKoshy, Sam M., Anthony E. Kincaid, and Jason C. Bartz. 2022. "Transport of Prions in the Peripheral Nervous System: Pathways, Cell Types, and Mechanisms" Viruses 14, no. 3: 630. https://doi.org/10.3390/v14030630