
NSG-Mice Reveal the Importance of a Functional Innate and Adaptive Immune Response to Overcome RVFV Infection

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus
2.2. Mice
2.3. Infection and Study Design
2.4. RNA Isolation and Reverse Transcription Quantitative Polymerase Chain Reaction
2.5. Serum Neutralization Test
2.6. Histology and Immunohistochemistry
2.7. Statistical Analysis
3. Results
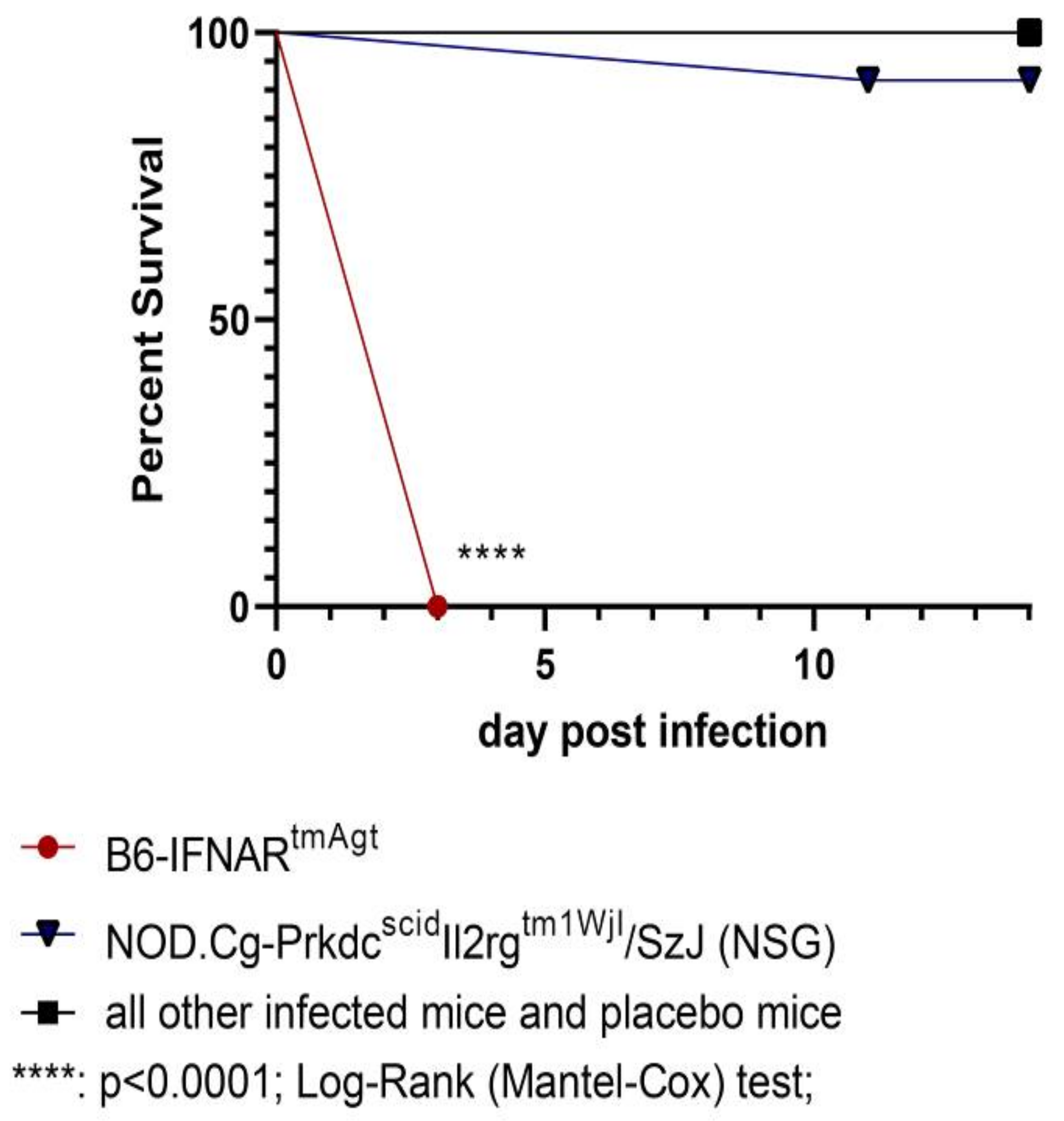
3.1. Survival and Clinical Signs
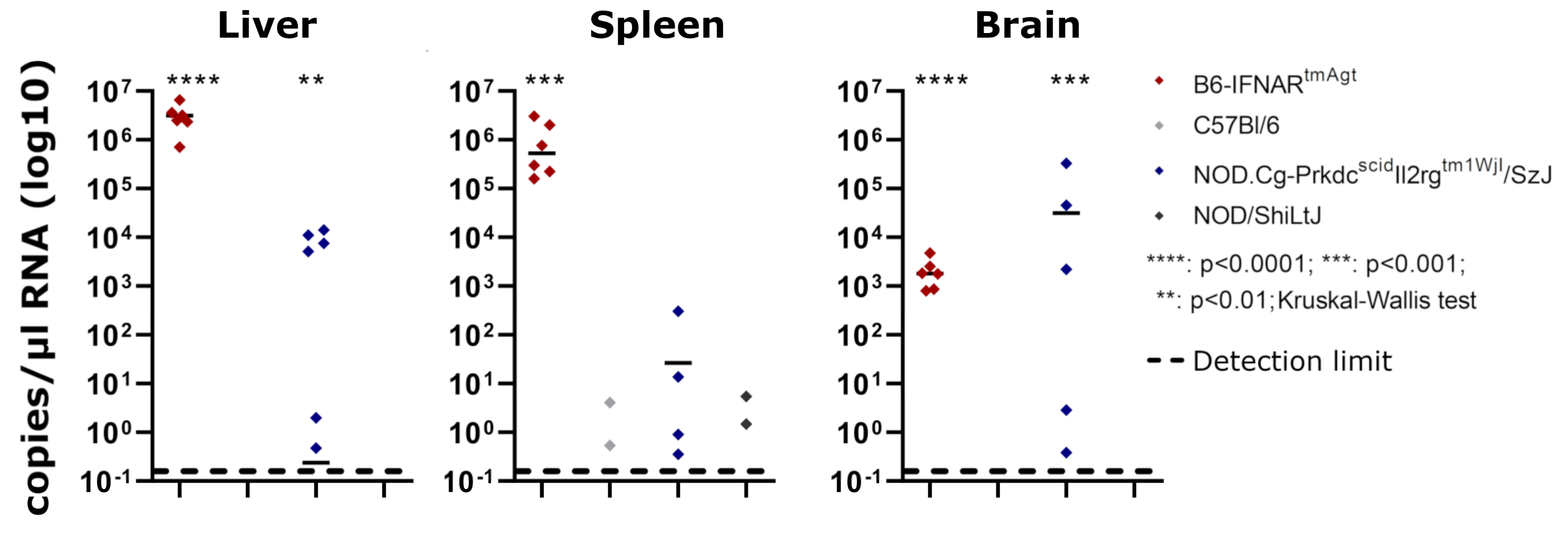
3.2. Reverse Transcription Quantitative Polymerase Chain Reaction
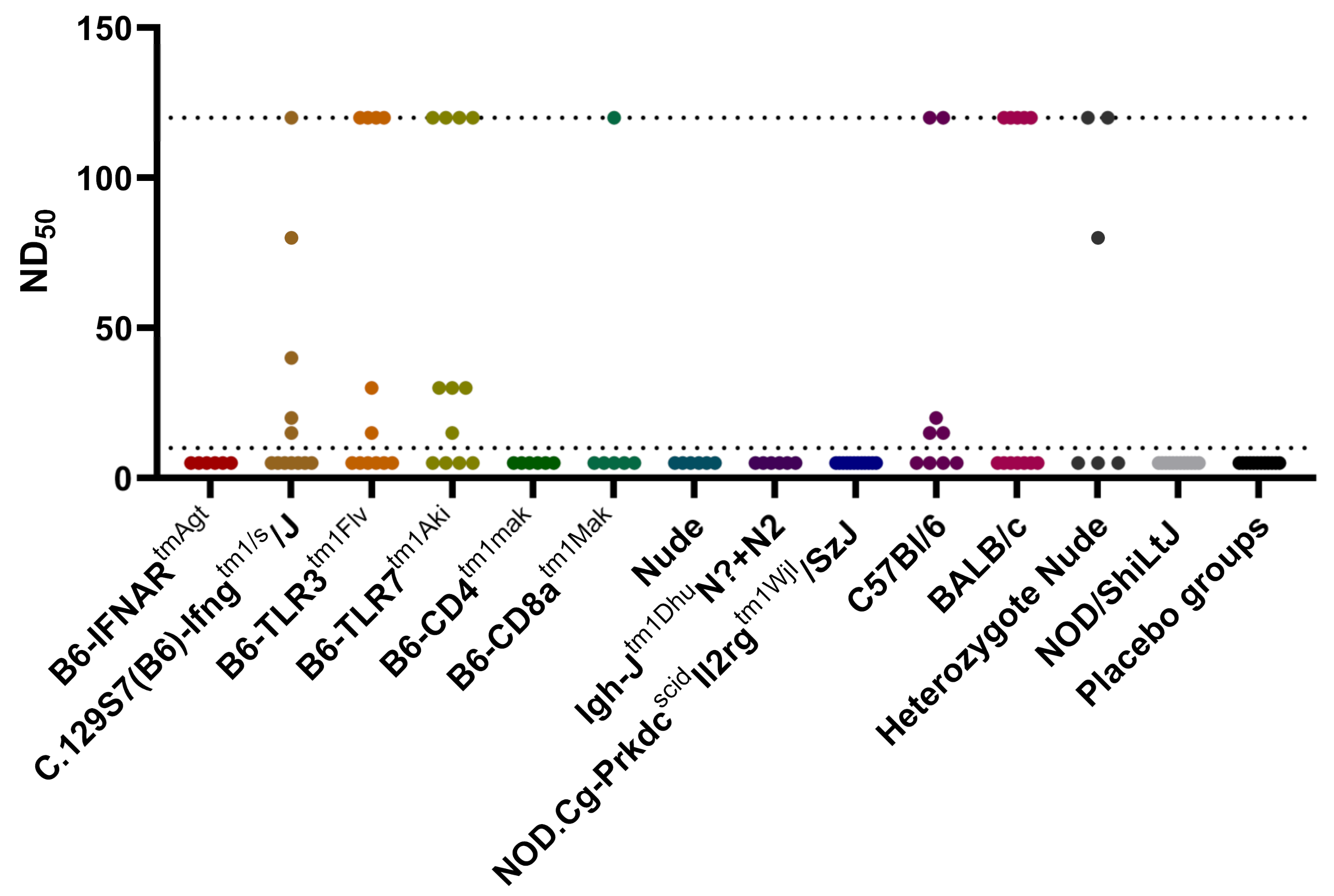
3.3. Serum Neutralization Test
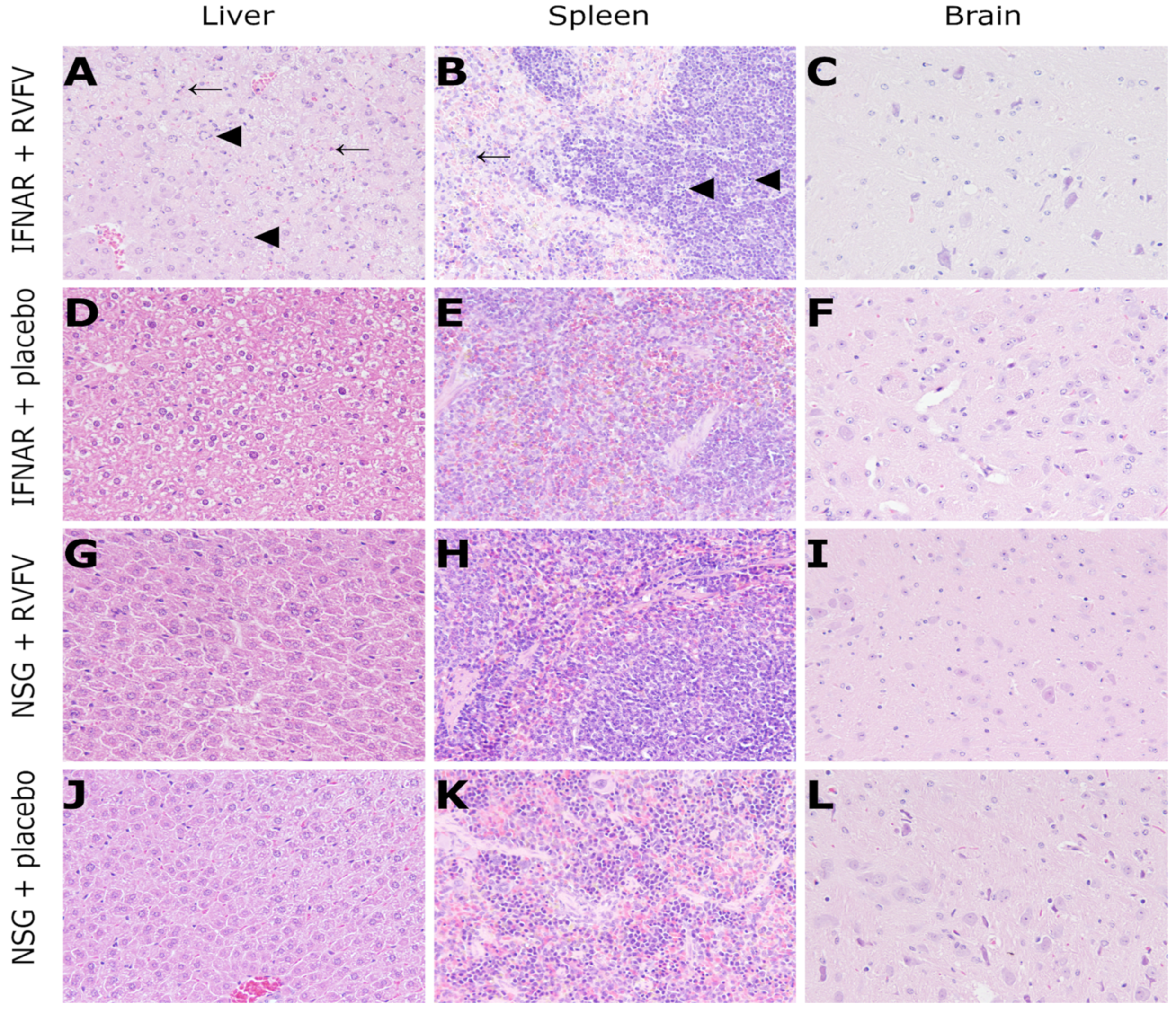
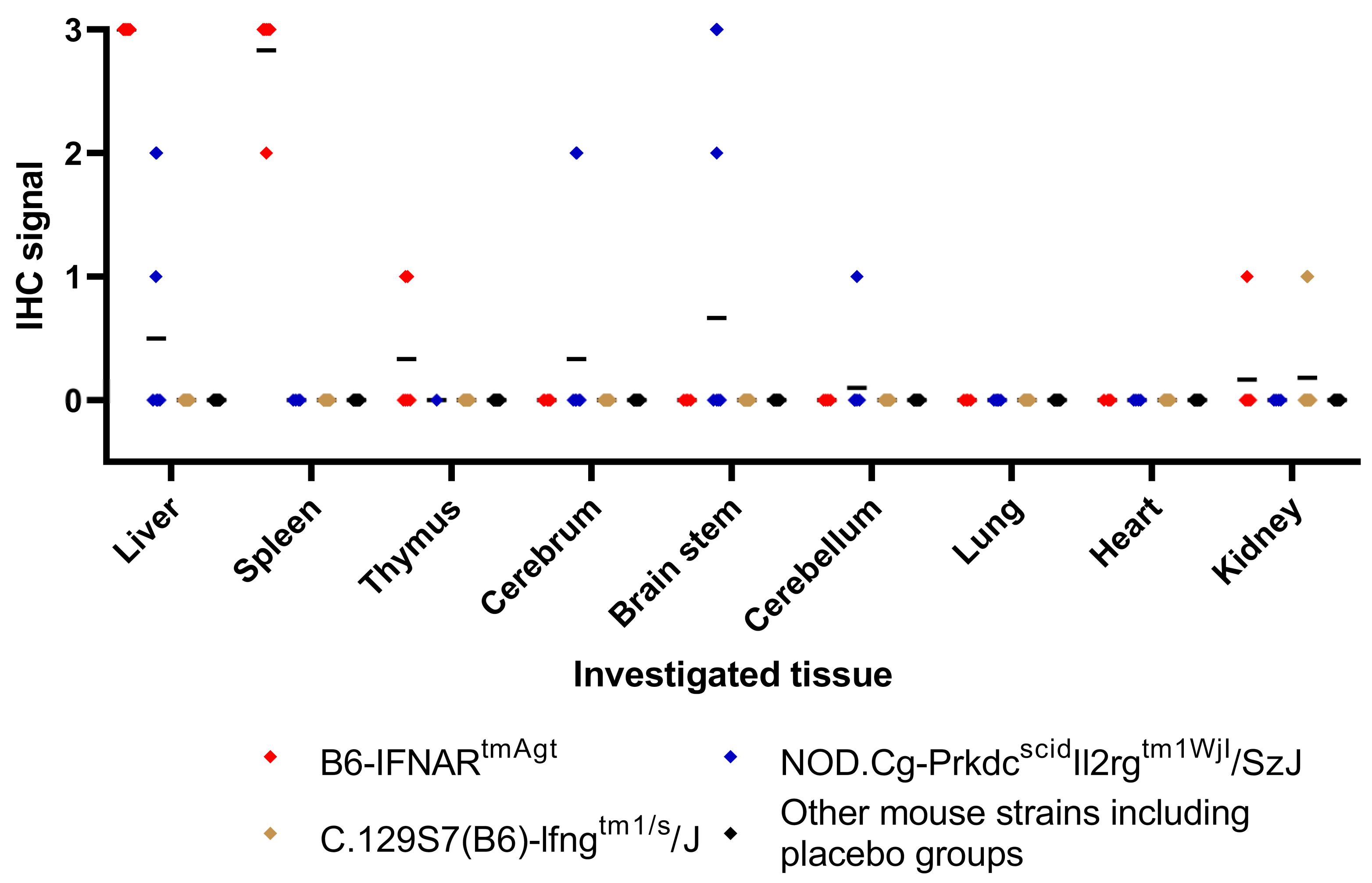
3.4. Histology and Immunohistochemistry
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Ulrich, R. Rift Valley Fever: An Ancient Plague on Its Way Out of Africa? Vet. Pathol. 2019, 56, 178–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, A. Rift Valley Fever. Clin. Lab. Med. 2017, 37, 285–301. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Makino, S. The pathogenesis of Rift Valley fever. Viruses 2011, 3, 493–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwaśnik, M.; Rożek, W.; Rola, J. Rift Valley Fever—A Growing Threat to Humans and Animals. J. Vet. Res. 2021, 65, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Muga, G.O.; Onyango-Ouma, W.; Sang, R.; Affognon, H. Sociocultural and economic dimensions of Rift Valley fever. Am. J. Trop. Med. Hyg 2015, 92, 730–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odendaal, L.; Davis, A.S.; Fosgate, G.T.; Clift, S.J. Lesions and Cellular Tropism of Natural Rift Valley Fever Virus Infection in Young Lambs. Vet. Pathol. 2020, 57, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Anyangu, A.S.; Gould, L.H.; Sharif, S.K.; Nguku, P.M.; Omolo, J.O.; Mutonga, D.; Rao, C.Y.; Lederman, E.R.; Schnabel, D.; Paweska, J.T.; et al. Risk factors for severe Rift Valley fever infection in Kenya, 2007. Am. J. Trop. Med. Hyg. 2010, 83, 14–21. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, B.M.; Russell, D.; dos Santos, I.; Gear, J.H. Rift Valley fever in humans in South Africa. S. Afr. Med. J. 1980, 58, 803–806. [Google Scholar] [PubMed]
- Pepin, M.; Bouloy, M.; Bird, B.H.; Kemp, A.; Paweska, J. Rift Valley fever virus (Bunyaviridae: Phlebovirus): An update on pathogenesis, molecular epidemiology, vectors, diagnostics and prevention. Vet. Res. 2010, 41, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinet, J.P.; Ferté, H.; Failloux, A.B.; Schaffner, F.; Depaquit, J. Mosquitoes of North-Western Europe as Potential Vectors of Arboviruses: A Review. Viruses 2019, 11, 1059. [Google Scholar] [CrossRef] [Green Version]
- OIE. OIE-Listed diseases, infections and infestations in force in 2021. Available online: https://www.oie.int/en/what-we-do/standards/codes-and-manuals/terrestrial-code-online-access/?id=169&L=0&htmfile=chapitre_rvf.htm (accessed on 28 April 2021).
- Nielsen, S.S.; Alvarez, J.; Bicout, D.J.; Calistri, P.; Depner, K.; Drewe, J.A.; Garin-Bastuji, B.; Rojas, J.L.G.; Schmidt, C.G.; Michel, V.; et al. Rift Valley Fever—Epidemiological update and risk of introduction into Europe. Efsa J. 2020, 18, e06041. [Google Scholar] [CrossRef] [Green Version]
- Bird, B.H.; McElroy, A.K. Rift Valley fever virus: Unanswered questions. Antiviral Res. 2016, 132, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Faburay, B.; LaBeaud, A.D.; McVey, D.S.; Wilson, W.C.; Richt, J.A. Current Status of Rift Valley Fever Vaccine Development. Vaccines 2017, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Kortekaas, J. One Health approach to Rift Valley fever vaccine development. Antiviral Res. 2014, 106, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Lerolle, S.; Freitas, N.; Cosset, F.L.; Legros, V. Host Cell Restriction Factors of Bunyaviruses and Viral Countermeasures. Viruses 2021, 13, 784. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, K.; Makino, S. Interplay between the Virus and Host in Rift Valley Fever Pathogenesis. J. Innate Immun. 2015, 7, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, G.; Lopez-Gil, E.; Warimwe, G.M.; Brun, A. Understanding Rift Valley fever: Contributions of animal models to disease characterization and control. Mol. Immunol. 2015, 66, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.M.; Bhardwaj, N.; Bissel, S.J.; Hartman, A.L.; Smith, D.R. Animal models of Rift Valley fever virus infection. Virus Res. 2012, 163, 417–423. [Google Scholar] [CrossRef]
- Lang, Y.; Henningson, J.; Jasperson, D.; Li, Y.; Lee, J.; Ma, J.; Li, Y.; Cao, N.; Liu, H.; Wilson, W.; et al. Mouse model for the Rift Valley fever virus MP12 strain infection. Vet. Microbiol. 2016, 195, 70–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.R.; Steele, K.E.; Shamblin, J.; Honko, A.; Johnson, J.; Reed, C.; Kennedy, M.; Chapman, J.L.; Hensley, L.E. The pathogenesis of Rift Valley fever virus in the mouse model. Virology 2010, 407, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Rissmann, M.; Kley, N.; Ulrich, R.; Stoek, F.; Balkema-Buschmann, A.; Eiden, M.; Groschup, M.H. Competency of Amphibians and Reptiles and Their Potential Role as Reservoir Hosts for Rift Valley Fever Virus. Viruses 2020, 12, 1206. [Google Scholar] [CrossRef]
- Gommet, C.; Billecocq, A.; Jouvion, G.; Hasan, M.; Zaverucha do Valle, T.; Guillemot, L.; Blanchet, C.; van Rooijen, N.; Montagutelli, X.; Bouloy, M.; et al. Tissue tropism and target cells of NSs-deleted rift valley fever virus in live immunodeficient mice. PLoS Negl. Trop. Dis. 2011, 5, e1421. [Google Scholar] [CrossRef] [Green Version]
- Odendaal, L.; Davis, A.S.; Venter, E.H. Insights into the Pathogenesis of Viral Haemorrhagic Fever Based on Virus Tropism and Tissue Lesions of Natural Rift Valley Fever. Viruses 2021, 13, 709. [Google Scholar] [CrossRef] [PubMed]
- Boyles, D.A.; Schwarz, M.M.; Albe, J.R.; McMillen, C.M.; O’Malley, K.J.; Reed, D.S.; Hartman, A.L. Development of Rift valley fever encephalitis in rats is mediated by early infection of olfactory epithelium and neuroinvasion across the cribriform plate. J. Gen. Virol. 2021, 102, 1522. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Weber, F. The interferon response circuit in antiviral host defense. Verh. K. Acad. Geneeskd. Belg. 2009, 71, 73–86. [Google Scholar] [PubMed]
- Lathan, R.; Simon-Chazottes, D.; Jouvion, G.; Godon, O.; Malissen, M.; Flamand, M.; Bruhns, P.; Panthier, J.J. Innate Immune Basis for Rift Valley Fever Susceptibility in Mouse Models. Sci. Rep. 2017, 7, 7096. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Semin. Immunol. 2007, 19, 24–32. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Renauld, J.C. Class II cytokine receptors and their ligands: Key antiviral and inflammatory modulators. Nat. Rev. Immunol. 2003, 3, 667–676. [Google Scholar] [CrossRef]
- Gowen, B.B.; Hoopes, J.D.; Wong, M.H.; Jung, K.H.; Isakson, K.C.; Alexopoulou, L.; Flavell, R.A.; Sidwell, R.W. TLR3 deletion limits mortality and disease severity due to Phlebovirus infection. J. Immunol. 2006, 177, 6301–6307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hise, A.G.; Traylor, Z.; Hall, N.B.; Sutherland, L.J.; Dahir, S.; Ermler, M.E.; Muiruri, S.; Muchiri, E.M.; Kazura, J.W.; LaBeaud, A.D.; et al. Association of symptoms and severity of rift valley fever with genetic polymorphisms in human innate immune pathways. PLoS Negl. Trop. Dis. 2015, 9, e0003584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElroy, A.K.; Harmon, J.R.; Flietstra, T.; Nichol, S.T.; Spiropoulou, C.F. Human Biomarkers of Outcome Following Rift Valley Fever Virus Infection. J. Infect. Dis. 2018, 218, 1847–1851. [Google Scholar] [CrossRef]
- Austin, D.; Baer, A.; Lundberg, L.; Shafagati, N.; Schoonmaker, A.; Narayanan, A.; Popova, T.; Panthier, J.J.; Kashanchi, F.; Bailey, C.; et al. p53 Activation following Rift Valley fever virus infection contributes to cell death and viral production. PLoS ONE 2012, 7, e36327. [Google Scholar] [CrossRef]
- Billecocq, A.; Spiegel, M.; Vialat, P.; Kohl, A.; Weber, F.; Bouloy, M.; Haller, O. NSs protein of Rift Valley fever virus blocks interferon production by inhibiting host gene transcription. J. Virol. 2004, 78, 9798–9806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouloy, M.; Janzen, C.; Vialat, P.; Khun, H.; Pavlovic, J.; Huerre, M.; Haller, O. Genetic evidence for an interferon-antagonistic function of rift valley fever virus nonstructural protein NSs. J. Virol. 2001, 75, 1371–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habjan, M.; Pichlmair, A.; Elliott, R.M.; Overby, A.K.; Glatter, T.; Gstaiger, M.; Superti-Furga, G.; Unger, H.; Weber, F. NSs protein of rift valley fever virus induces the specific degradation of the double-stranded RNA-dependent protein kinase. J. Virol. 2009, 83, 4365–4375. [Google Scholar] [CrossRef] [Green Version]
- Kalveram, B.; Lihoradova, O.; Indran, S.V.; Lokugamage, N.; Head, J.A.; Ikegami, T. Rift Valley fever virus NSs inhibits host transcription independently of the degradation of dsRNA-dependent protein kinase PKR. Virology 2013, 435, 415–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansuroglu, Z.; Josse, T.; Gilleron, J.; Billecocq, A.; Leger, P.; Bouloy, M.; Bonnefoy, E. Nonstructural NSs protein of rift valley fever virus interacts with pericentromeric DNA sequences of the host cell, inducing chromosome cohesion and segregation defects. J. Virol. 2010, 84, 928–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Struthers, J.K.; Swanepoel, R. Identification of a major non-structural protein in the nuclei of Rift Valley fever virus-infected cells. J. Gen. Virol. 1982, 60, 381–384. [Google Scholar] [CrossRef]
- Won, S.; Ikegami, T.; Peters, C.J.; Makino, S. NSm protein of Rift Valley fever virus suppresses virus-induced apoptosis. J. Virol. 2007, 81, 13335–13345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenborn, J.R.; Wilson, C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar] [CrossRef]
- Harmon, J.R.; Spengler, J.R.; Coleman-McCray, J.D.; Nichol, S.T.; Spiropoulou, C.F.; McElroy, A.K. CD4 T Cells, CD8 T Cells, and Monocytes Coordinate To Prevent Rift Valley Fever Virus Encephalitis. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Wright, D.; Kortekaas, J.; Bowden, T.A.; Warimwe, G.M. Rift Valley fever: Biology and epidemiology. J. Gen. Virol. 2019, 100, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Watts, D.M.; Costanzo, M.C.; Tang, X.; Venegas, L.A.; Jiao, F.; Sette, A.; Sidney, J.; Sewell, A.K.; Wooldridge, L.; et al. The nucleocapsid protein of Rift Valley fever virus is a potent human CD8+ T cell antigen and elicits memory responses. PLoS ONE 2013, 8, e59210. [Google Scholar] [CrossRef]
- Gregor, K.M.; Michaely, L.M.; Gutjahr, B.; Rissmann, M.; Keller, M.; Dornbusch, S.; Naccache, F.; Schön, K.; Jansen, S.; Heitmann, A.; et al. Rift Valley fever virus detection in susceptible hosts with special emphasis in insects. Sci. Rep. 2021, 11, 9822. [Google Scholar] [CrossRef]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn-Schmiedebergs Archiv für Experimentelle Pathologie und Pharmakologie 1931, 162, 480–483. [Google Scholar] [CrossRef]
- SPEARMAN, C. The method of right and wrong cases (constant stimuli) without Gauss’s formulae. Br. J. Psychol. 1908, 2, 227–242. [Google Scholar] [CrossRef]
- Müller, U.; Steinhoff, U.; Reis, L.F.L.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional Role of Type I and Type II Interferons in Antiviral Defense. Science 1994, 264, 1918–1921. [Google Scholar] [CrossRef]
- Dalton, D.K.; Pitts-Meek, S.; Keshav, S.; Figari, I.S.; Bradley, A.; Stewart, T.A. Multiple Defects of Immune Cell Function in Mice with Disrupted Interferon-γ Genes. Science 1993, 259, 1739–1742. [Google Scholar] [CrossRef]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small anti-viral compounds activate immune cells via the TLR7 MyD88–dependent signaling pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Rahemtulla, A.; Fung-Leung, W.; Schilham, M.; Kündig, T.; Sambhara, S.; Narendran, A.; Arabian, A.; Wakeham, A.; Paige, C.; Zinkernagel, R. Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature 1991, 353, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, A.M.; Harmon, C.; Whelan, S.; O’Brien, E.C.; O’Reilly, V.P.; Crotty, P.; Kelly, P.; Ryan, M.; Hickey, F.B.; O’Farrelly, C.; et al. Myeloid Engraftment in Humanized Mice: Impact of Granulocyte-Colony Stimulating Factor Treatment and Transgenic Mouse Strain. Stem Cells Dev. 2016, 25, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Pantelouris, E.M. Absence of thymus in a mouse mutant. Nature 1968, 217, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Longacre, A.; Meng, F.; Patel, V.; Hsiao, K.; Koh, J.S.; Levine, J.S. Cytokine dysregulation induced by apoptotic cells is a shared characteristic of macrophages from nonobese diabetic and systemic lupus erythematosus-prone mice. J. Immunol. 2004, 172, 4834–4843. [Google Scholar] [CrossRef] [Green Version]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, S.; Satoh, J.; Fujiya, H.; Toyota, T.; Suzuki, R.; Itoh, K.; Kumagai, K. Immunologic aspects of the nonobese diabetic (NOD) mouse. Abnormalities of cellular immunity. Diabetes 1983, 32, 247–253. [Google Scholar] [CrossRef]
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.V.; Greiner, D.L.; et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995, 154, 180–191. [Google Scholar]
- Leiter, E.H. The NOD mouse: A model for analyzing the interplay between heredity and environment in development of autoimmune disease. ILAR J. 1993, 35, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Glueck, D. Sample Size Calculations in Clinical Research, 2nd ed.; Chow, S.-C., Shao, J., Wang, H., Eds.; CRC Press: Boca Raton, FL, USA, 2008; Chapter 7.3; p. 180. [Google Scholar]
- Schoenfeld, D.A. Sample-size formula for the proportional-hazards regression model. Biometrics 1983, 39, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Ninove, L.; Nougairede, A.; Gazin, C.; Thirion, L.; Delogu, I.; Zandotti, C.; Charrel, R.N.; De Lamballerie, X. RNA and DNA bacteriophages as molecular diagnosis controls in clinical virology: A comprehensive study of more than 45,000 routine PCR tests. PLoS ONE 2011, 6, e16142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, B.H.; Bawiec, D.A.; Ksiazek, T.G.; Shoemaker, T.R.; Nichol, S.T. Highly sensitive and broadly reactive quantitative reverse transcription-PCR assay for high-throughput detection of Rift Valley fever virus. J. Clin. Microbiol. 2007, 45, 3506–3513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäckel, S.; Eiden, M.; El Mamy, B.O.; Isselmou, K.; Vina-Rodriguez, A.; Doumbia, B.; Groschup, M.H. Molecular and serological studies on the Rift Valley fever outbreak in Mauritania in 2010. Transbound Emerg. Dis. 2013, 60 (Suppl. 2), 31–39. [Google Scholar] [CrossRef] [PubMed]
- Rissmann, M.; Ulrich, R.; Schroder, C.; Hammerschmidt, B.; Hanke, D.; Mroz, C.; Groschup, M.H.; Eiden, M. Vaccination of alpacas against Rift Valley fever virus: Safety, immunogenicity and pathogenicity of MP-12 vaccine. Vaccine 2017, 35, 655–662. [Google Scholar] [CrossRef] [PubMed]
- RITA. Revised guides for organ sampling and trimming in rats and mice. Available online: https://reni.item.fraunhofer.de/reni/trimming/index.php (accessed on 3 February 2022).
- Fischer, A.H.; Jacobson, K.A.; Rose, J.; Zeller, R. Hematoxylin and eosin staining of tissue and cell sections. CSH Protoc. 2008, 2008, pdb-prot4986. [Google Scholar] [CrossRef] [PubMed]
- Stoek, F.; Rissmann, M.; Ulrich, R.; Eiden, M.; Groschup, M.H. Black rats (Rattus rattus) as potential reservoir hosts for Rift Valley fever phlebovirus: Experimental infection results in viral replication and shedding without clinical manifestation. Transbound. Emerg. Dis. 2021, 00, 1–12. [Google Scholar] [CrossRef]
- Aoshi, T.; Koyama, S.; Kobiyama, K.; Akira, S.; Ishii, K.J. Innate and adaptive immune responses to viral infection and vaccination. Curr. Opin. Virol. 2011, 1, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Reed, C.; Steele, K.E.; Honko, A.; Shamblin, J.; Hensley, L.E.; Smith, D.R. Ultrastructural study of Rift Valley fever virus in the mouse model. Virology 2012, 431, 58–70. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Prakash, S.; Garg, R.K.; Jain, P.; Kumar, R.; Jain, A. Study of Single Nucleotide Polymorphisms in Endosomal Toll-Like Receptors-3, 7, and 9 Genes in Patients With Dengue: A Case-Control Study. Cureus 2021, 13, e14883. [Google Scholar] [CrossRef] [PubMed]
- Pérez de Diego, R.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; Plancoulaine, S.; Picard, C.; Herman, M.; Cardon, A.; Durandy, A.; Bustamante, J.; et al. Human TRAF3 Adaptor Molecule Deficiency Leads to Impaired Toll-like Receptor 3 Response and Susceptibility to Herpes Simplex Encephalitis. Immunity 2010, 33, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Cader, M.S.; De Silva Senapathi, U.; Nagy, E.; Sharif, S.; Abdul-Careem, M.F. Antiviral response elicited against avian influenza virus infection following activation of toll-like receptor (TLR)7 signaling pathway is attributable to interleukin (IL)-1β production. BMC Res. Notes 2018, 11, 859. [Google Scholar] [CrossRef] [PubMed]
- Wichgers Schreur, P.J.; Kant, J.; van Keulen, L.; Moormann, R.J.; Kortekaas, J. Four-segmented Rift Valley fever virus induces sterile immunity in sheep after a single vaccination. Vaccine 2015, 33, 1459–1464. [Google Scholar] [CrossRef]
- Dodd, K.A.; McElroy, A.K.; Jones, M.E.; Nichol, S.T.; Spiropoulou, C.F. Rift Valley fever virus clearance and protection from neurologic disease are dependent on CD4+ T cell and virus-specific antibody responses. J. Virol. 2013, 87, 6161–6171. [Google Scholar] [CrossRef] [Green Version]
- Faburay, B.; Wilson, W.C.; Gaudreault, N.N.; Davis, A.S.; Shivanna, V.; Bawa, B.; Sunwoo, S.Y.; Ma, W.; Drolet, B.S.; Morozov, I.; et al. A Recombinant Rift Valley Fever Virus Glycoprotein Subunit Vaccine Confers Full Protection against Rift Valley Fever Challenge in Sheep. Sci. Rep. 2016, 6, 27719. [Google Scholar] [CrossRef] [PubMed]
- McElroy, A.K.; Nichol, S.T. Rift Valley fever virus inhibits a pro-inflammatory response in experimentally infected human monocyte derived macrophages and a pro-inflammatory cytokine response may be associated with patient survival during natural infection. Virology 2012, 422, 6–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, K.K.; Hill, T.E.; Davis, M.N.; Holbrook, M.R.; Freiberg, A.N. Cytokine response in mouse bone marrow derived macrophages after infection with pathogenic and non-pathogenic Rift Valley fever virus. J. Gen. Virol. 2015, 96, 1651–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Jackson Laboratory. NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ. Available online: https://www.jax.org/strain/005557 (accessed on 3 February 2022).
- Brehm, M.A.; Wiles, M.V.; Greiner, D.L.; Shultz, L.D. Generation of improved humanized mouse models for human infectious diseases. J. Immunol. Methods 2014, 410, 3–17. [Google Scholar] [CrossRef]
- Ma, L.; Li, Q.; Cai, S.; Peng, H.; Huyan, T.; Yang, H. The role of NK cells in fighting the virus infection and sepsis. Int. J. Med. Sci. 2021, 18, 3236–3248. [Google Scholar] [CrossRef]
- Braun, M.; Björkström, N.K.; Gupta, S.; Sundström, K.; Ahlm, C.; Klingström, J.; Ljunggren, H.G. NK cell activation in human hantavirus infection explained by virus-induced IL-15/IL15Rα expression. PLoS Pathog. 2014, 10, e1004521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingström, J.; Smed-Sörensen, A.; Maleki, K.T.; Solà-Riera, C.; Ahlm, C.; Björkström, N.K.; Ljunggren, H.G. Innate and adaptive immune responses against human Puumala virus infection: Immunopathogenesis and suggestions for novel treatment strategies for severe hantavirus-associated syndromes. J. Intern. Med. 2019, 285, 510–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, N.; Brown, S. The mouse ascending: Perspectives for human-disease models. Nat. Cell Biol. 2007, 9, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas, J.; Oreshkova, N.; van Keulen, L.; Kant, J.; Bosch, B.J.; Bouloy, M.; Moulin, V.; Goovaerts, D.; Moormann, R.J. Comparative efficacy of two next-generation Rift Valley fever vaccines. Vaccine 2014, 32, 4901–4908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplen, H.; Peters, C.J.; Bishop, D.H. Mutagen-directed attenuation of Rift Valley fever virus as a method for vaccine development. J. Gen. Virol. 1985, 66, 2271–2277. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Hill, T.E.; Smith, J.K.; Zhang, L.; Juelich, T.L.; Gong, B.; Slack, O.A.; Ly, H.J.; Lokugamage, N.; Freiberg, A.N. Rift Valley Fever Virus MP-12 Vaccine Is Fully Attenuated by a Combination of Partial Attenuations in the S, M, and L Segments. J. Virol. 2015, 89, 7262–7276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrill, J.C.; Jennings, G.B.; Caplen, H.; Turell, M.J.; Johnson, A.J.; Peters, C.J. Pathogenicity and immunogenicity of a mutagen-attenuated Rift Valley fever virus immunogen in pregnant ewes. Am. J. Vet. Res. 1987, 48, 1042–1047. [Google Scholar]
- Morrill, J.C.; Peters, C.J. Protection of MP-12-vaccinated rhesus macaques against parenteral and aerosol challenge with virulent rift valley fever virus. J. Infect. Dis 2011, 204, 229–236. [Google Scholar] [CrossRef]
- Bartlett, B.L.; Pellicane, A.J.; Tyring, S.K. Vaccine immunology. Dermatol. Ther. 2009, 22, 104–109. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Designation | Immunological Trait | Number of Infected/Placebo Animals | Origin | References |
|---|---|---|---|---|---|
| 1 | B6-IFNARtmAgt | No IFNAR expression | 6/6 | FLI | [50] |
| 2 | C.129S7(B6)-Ifngtm1/s/J | Impaired Interferon γ response and therefore decreased activity of macrophage function | 12/6 | FLI | [51] |
| 3 | B6-TLR3tm1Flv | TLR 3 deficiency | 12/6 | FLI | [52] |
| 4 | B6-TLR7tm1Aki | TLR 7 deficiency | 12/6 | FLI | [52] |
| 5 | B6-CD4tm1Mak | Block of CD4+ T-Lymphocyte development and restricted T-helper-cell response | 6/6 | FLI | [53] |
| 6 | B6-CD8atm1Mak | Deficient in functional cytotoxic T-Lymphocytes | 6/6 | FLI | [54] |
| 7 | Foxn1nu-/- (NUDE) | Lack of thymus and therefore absence of T-Lymphocytes, partial defect in B-cell development | 6/6 | JAX | [55] |
| 8 | Igh-Jtm1DhuN?+N2 | No B-cell maturation, therefore no IgM or IgG production | 6/6 | TAC | [56] |
| 9 | NOD.Cg-Prkdcscid Il2rgtm1WjI/SzJ (NSG) | No lymphocyte maturation (B and T cells), therefore no IgG and extremely low cytotoxic T-cells, deficiency of NK cells, macrophages and dendritic cells, absence of complement C5 | 12/6 | CRL | [54,56,57,58,59] |
| 10 | C57Bl/6 | Wildtype, background of strain 1, 3-6 | 9/6 | FLI | [20] |
| 11 | BALB/c | Wildtype, background of strain 2 and 8 | 12/6 | FLI | [20] |
| 12 | Foxn1nu+/- (NUDE heterozygous) | Heterozygous control for strain 7 | 6/6 | JAX | [55] |
| 13 | NOD/ShiLtJ (NOD) | Background of strain 9 (NSG), late-onset spontaneous Autoimmune diabetes mellitus, deficient NK cells, macrophages, dendritic cells and complement component C5 | 12/6 | CRL | [56,58,59,60] |
| Category | Description of Signs | Score |
|---|---|---|
| Posture and appearance | Normal posture, smooth fur | 0 |
| Normal posture, ruffled fur | 1 | |
| Mildly hunched back, ruffled fur | 2 | |
| Severely hunched back, ruffled fur, lack of cleaning | 3 | |
| Behavior and activity | Curious and alert | 0 |
| Calm, mildly reduced spontaneous movement | 1 | |
| Apathy, moderately reduced spontaneous movement, mildly reduced induced movement | 2 | |
| Stupor, no spontaneous movement, severely reduced induced movement | 3 | |
| Body weight | No change >5% | 0 |
| Decrease of 5–15% | 1 | |
| Decrease of 15–25% | 2 | |
| Decrease of >25% | 3 |
| Mouse Strain | Liver | Spleen | Brain |
|---|---|---|---|
| B6-IFNARtmAgt | 6/6 (7.08 × 105–6.54 × 106) | 6/6 (1.6 × 105–3.02 × 106) | 6/6 (8.03 × 102–4.76 × 103) |
| C.129S7(B6)-Ifngtm1/s/J | n.d. | 3/12 (4.4 × 10−1–2.54 × 101) | n.d. |
| B6-TLR3tm1Flv | n.d. | n.d. | n.d. |
| B6-TLR7tm1Aki | n.d. | 5/12 (6.28 × 10−1–5.94 × 100) | n.d. |
| B6-CD4tm1Mak | n.d. | n.d. | n.d. |
| B6-CD8atm1Mak | n.d. | 1/6 (1 × 102) | n.d. |
| Foxn1nu-/- (NUDE) | n.d. | n.d. | n.d. |
| Igh-Jtm1DhuN?+N2 | 2/6 (4.08 × 10−3–8.38 × 10−2) | n.d. | n.d. |
| NOD.Cg-Prkdcscid Il2rgtm1WjI/SzJ (NSG) | 6/12 (4.74 × 10−1–1.41 × 104) | 4/12 (3.53 × 10−1–3.03 × 102) | 6/12 (2.87 × 10−2–3.29 × 105) |
| C57Bl/6 | n.d. | 2/9 (5.41 × 10−1–4.06 × 100) | 1/9 (2.37 × 101) |
| BALB/c | n.d. | 3/12 (1.41 × 100–7.05 × 100) | n.d. |
| Foxn1nu+/- (heterozygous NUDE) | n.d. | n.d. | n.d. |
| NOD/ShiLtJ (NOD) | n.d. | 2/12 (1.47 × 100–5.43 × 100) | n.d. |
| Mouse Strain | Histology | IHC Liver | IHC Spleen | IHC Brain | IHC Heart | IHC Lung | IHC Kidney |
|---|---|---|---|---|---|---|---|
| B6-IFNARtmAgt | Severe, diffuse, hepatocellular necrosis and apoptosis; Mild to moderate lymphocytolysis in the spleen | +++ | +++ | + (3/6) - (3/6) | - | + (1/6) -(5/6) | +(2/6) -(4/6) |
| NOD.Cg-Prkdcscid Il2rgtm1WjI/SzJ (NSG) | Mild, multifocal, hepatocellular necrosis and apoptosis (2/12); No findings (10/12) | ++ | - | +++ | - | +(2/12) -(10/12) | ++(3/12) -(9/12) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michaely, L.M.; Rissmann, M.; Keller, M.; König, R.; von Arnim, F.; Eiden, M.; Rohn, K.; Baumgärtner, W.; Groschup, M.; Ulrich, R. NSG-Mice Reveal the Importance of a Functional Innate and Adaptive Immune Response to Overcome RVFV Infection. Viruses 2022, 14, 350. https://doi.org/10.3390/v14020350
Michaely LM, Rissmann M, Keller M, König R, von Arnim F, Eiden M, Rohn K, Baumgärtner W, Groschup M, Ulrich R. NSG-Mice Reveal the Importance of a Functional Innate and Adaptive Immune Response to Overcome RVFV Infection. Viruses. 2022; 14(2):350. https://doi.org/10.3390/v14020350
Chicago/Turabian StyleMichaely, Lukas Mathias, Melanie Rissmann, Markus Keller, Rebecca König, Felicitas von Arnim, Martin Eiden, Karl Rohn, Wolfgang Baumgärtner, Martin Groschup, and Reiner Ulrich. 2022. "NSG-Mice Reveal the Importance of a Functional Innate and Adaptive Immune Response to Overcome RVFV Infection" Viruses 14, no. 2: 350. https://doi.org/10.3390/v14020350