
Alphacoronaviruses Are Common in Bats in the Upper Midwestern United States


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection and Processing
2.3. RNA Extractions
2.4. Nested Pan-CoV PCR
2.5. Sanger Sequencing
2.6. Metagenomic Sequencing
2.7. Phylogenetic Analysis
2.8. Recombination Analysis
2.9. Viral Isolation
2.10. Accession Numbers
3. Results
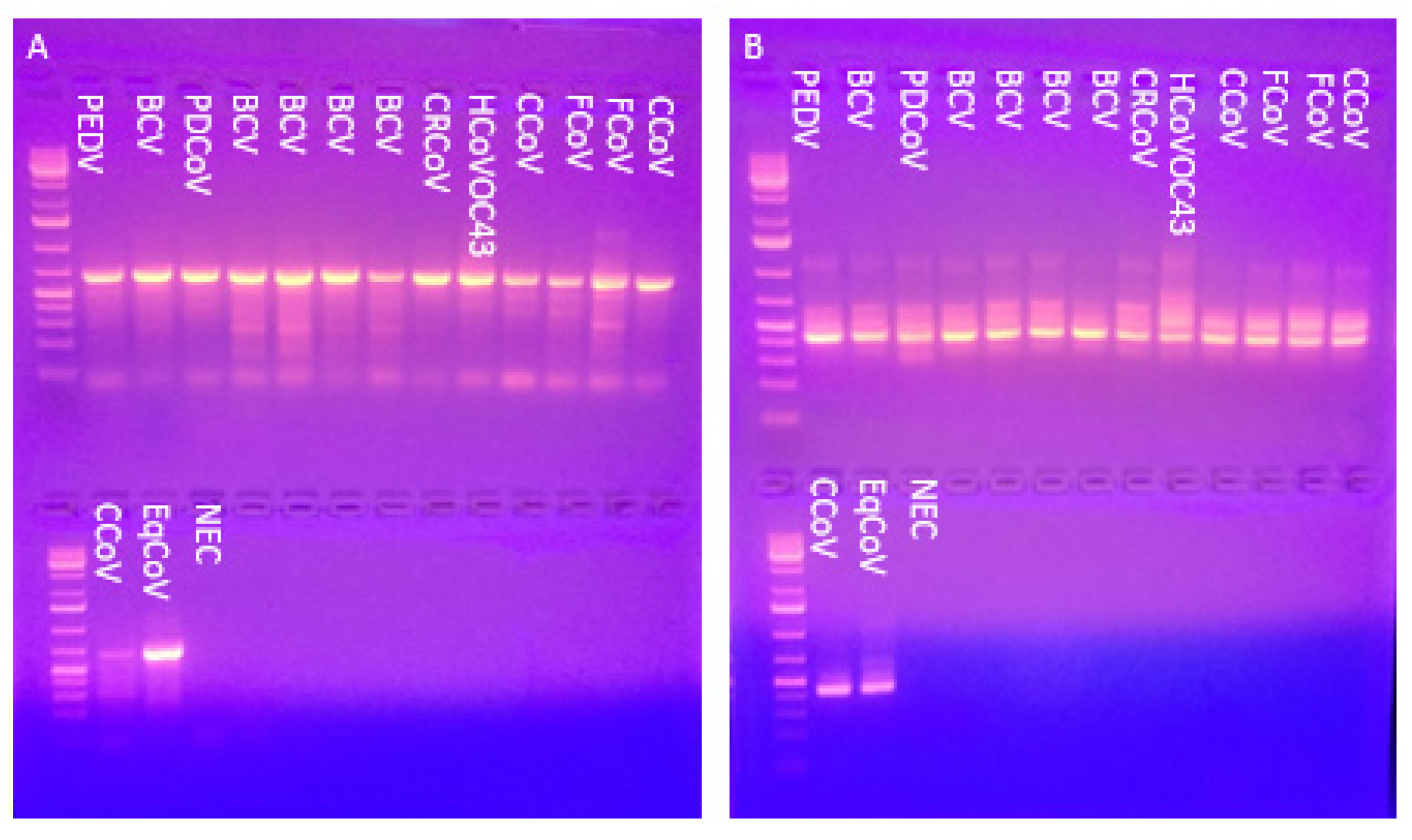
3.1. The Pan-Coronavirus PCR Detected All 15 Reference Coronaviruses
3.2. Novel Alphacoronavirus Discovered in Bats from South Dakota and Minnesota
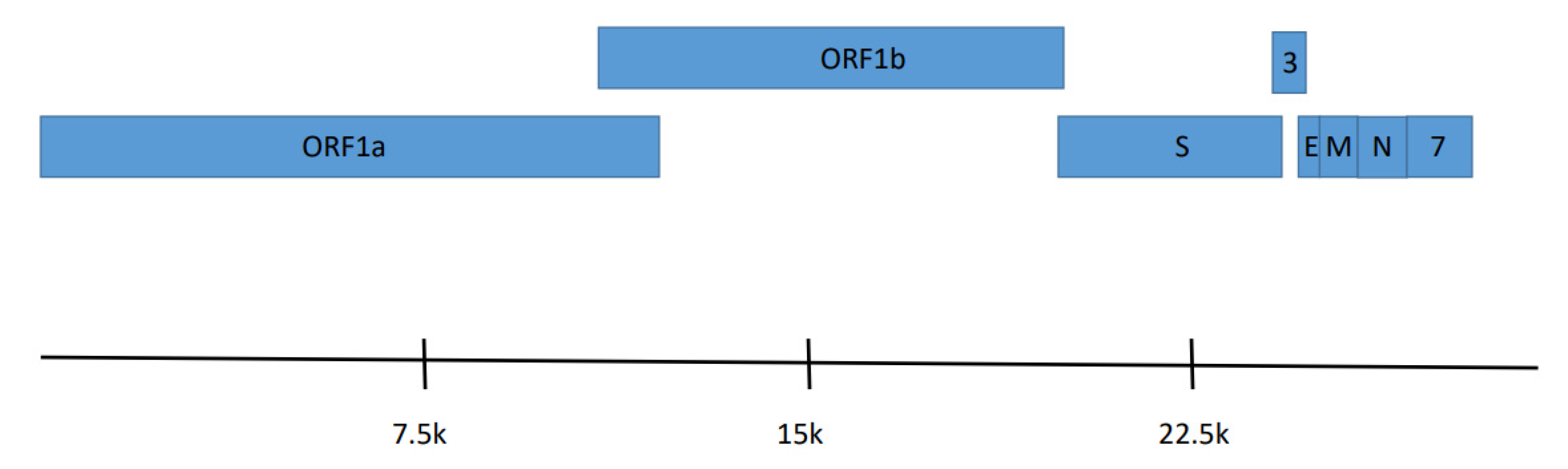
3.3. Genome Features of Eptesicus Bat Coronavirus
3.4. EbCoV Proteins Are Most Similar to Bat and Porcine Alphacoronaviruses
3.5. Eptesicus Bat Coronavirus Is Most Closely Related to Bat CoV HKU2
3.6. Recombination Analysis
3.7. Virus Isolation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Dempsey, D.M.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Hendrickson, R.C.; et al. Changes to virus taxonomy and the Statutes ratified by the International Committee on Taxonomy of Viruses (2020). Arch. Virol. 2020, 165, 2737–2748. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.X.; Ng, Y.L.; Tam, J.P.; Liu, D.X. Human Coronaviruses: A Review of Virus-Host Interactions. Diseases 2016, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Bio. 2016, 1282, 1–23. [Google Scholar]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol. 2012, 86, 3995–4008. [Google Scholar] [PubMed] [Green Version]
- Van der Hoek, L.; Pyrc, K.; Berkhout, B. Human coronavirus NL63, a new respiratory virus. FEMS Microbiol Rev. 2006, 30, 760–773. [Google Scholar] [CrossRef] [Green Version]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Hoek, L. Human coronaviruses: What do they cause? Antivir. Ther. 2007, 12, 651–658. [Google Scholar]
- Woo, P.C.; Lau, S.K.; Yip, C.C.; Huang, Y.; Yuen, K.Y. More and More Coronaviruses: Human Coronavirus HKU1. Viruses 2009, 1, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Schoeman, D.; Fielding, B.C. Human Coronaviruses: Counteracting the Damage by Storm. Viruses 2007, 13, 1457. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today 2020, 25, 668–688. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Ge, X.; Wang, L.F.; Shi, Z. Bat origin of human coronaviruses. Virol. J. 2015, 12, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitner, T.; Kumar, S. Where Did SARS-CoV-2 Come From? Mol. Biol. Evol. 2020, 37, 2463–2464. [Google Scholar] [CrossRef]
- Dominguez, S.R.; O’Shea, T.J.; Oko, L.M.; Holmes, K.V. Detection of group 1 coronaviruses in bats in North America. Emerg. Infect. Dis. 2007, 13, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, E.F.; Haskew, A.N.; Gates, J.E.; Huynh, J.; Moore, C.J.; Frieman, M.B. Metagenomic analysis of the viromes of three North American bat species: Viral diversity among different bat species that share a common habitat. J. Virol. 2010, 84, 13004–13018. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Aguilar, I.; Lorenzo, C.; Santos-Moreno, A.; Naranjo, E.; Navarrete-Gutierrez, D. Coronaviruses in bats: A review for the Americans. Viruses 2021, 13, 1226. [Google Scholar] [CrossRef]
- Hu, H.; Jung, K.; Wang, Q.; Saif, L.J.; Vlasova, A.N. Development of a one-step RT-PCR assay for detection of pancoronaviruses (α-, β-, γ-, and δ-coronaviruses) using newly designed degenerate primers for porcine and avian ‘fecal samples. J. Viro. Met. 2018, 256, 116–122. [Google Scholar] [CrossRef]
- Holbrook, M.G.; Anthony, S.J.; Navarrete-Macias, I.; Bestebroer, T.; Munster, V.J.; van Doremalen, N. Updated and Validated PanCoronavirus PCR Assay to Detect All Coronavirus Genera. Viruses 2021, 13, 599. [Google Scholar]
- Djikeng, A.; Halpin, R.; Kuzmickas, R.; De Passe, J.; Feldblyum, J.; Sengamalay, N.; Afonso, C.; Zhang, X.; Anderson, N.G.; Ghedin, E.; et al. 2008. Viral genome sequencing by random priming methods. BMC Genom. 2008, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, H.Q.; Nguyen, V.G.; Chung, C.U.; Jeon, Y.S.; Shin, S.; Jang, K.C.; Pham, L.B.H.; Kong, A.; Kim, C.U.; Park, Y.H.; et al. Genomic characterization of a novel alphacoronavirus isolated from bats, Korea, 2020. Viruses 2021, 13, 2041. [Google Scholar] [CrossRef]
- Chen, L.L.; Ou, H.Y.; Zhang, R.; Zhang, C.T. ZCURVE_CoV: A new system to recognize protein coding genes in coronavirus genomes, and its applications in analyzing SARS-CoV genomes. Biochem. Biophys. Res. Commun. 2003, 307, 382–388. [Google Scholar] [CrossRef]
- Koopman, K.F. Order Chiroptera. In Mammal Species of the World; Wilson, D.E., Reeder, D.M., Eds.; Smithsonian Institute Press: Washington, DC, USA, 1993; pp. 137–241. [Google Scholar]
- Hause, B.M.; Nelson, E.; Henning, C. Eptesicus fuscus orthorubulavirus, a close relative of human parainfluenza virus 4, discovered in a bat in South Dakota. Microbiol. Spectr. 2021, 9, e0093021. [Google Scholar] [CrossRef] [PubMed]
- Hause, B.M.; Nelson, E.; Christopher-Hennings, J. Novel and diverse non-rabies Rhabdoviruses identified in bats with human exposure, South Dakota, USA. Viruses 2020, 12, 1408. [Google Scholar] [CrossRef]
- Subudhi, S.; Rapin, N.; Dorville, N.; Hill, J.E.; Town, J.; Willis, C.K.R.; Bollinger, T.K.; Misra, V. Isolation, characterization and prevalence of a novel gammaherpesvirus in Eptesicus fuscus, the North American big brown bat. Virology 2018, 516, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Emerson, G.L.; Nordhausen, R.; Garner, M.M.; Huckabee, J.R.; Johnson, S.; Wohrle, R.D.; Davidson, W.B.; Wilkins, K.; Li, Y.; Doty, J.B.; et al. Novel poxvirus in big brown bats, northwestern United States. Emerg. Infect. Dis. 2013, 19, 1002–1004. [Google Scholar] [CrossRef] [PubMed]
- George, D.B.; Webb, C.T.; Farnsworth, M.L.; O’Shea, T.J.; Bowen, R.A.; Smith, D.L.; Stanley, T.R.; Ellison, L.E.; Rupprecht, C.E. Host and viral ecology determine bat rabies seasonality and maintenance. Proc. Natl. Acad. Sci. USA 2011, 108, 10208–10213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.L.; Shi, W.F.; Zhang, W.; Zhu, Y.; Zhang, Y.W.; Xie, Q.M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef]
- Yang, Y.L.; Qin, P.; Wang, B.; Liu, Y.; Xu, G.H.; Peng, L.; Zhou, J.; Zhu, S.J.; Huang, Y.W. Broad Cross-Species Infection of Cultured Cells by Bat HKU2-Related Swine Acute Diarrhea Syndrome Coronavirus and Identification of Its Replication in Murine Dendritic Cells In Vivo Highlight Its Potential for Diverse Interspecies Transmission. J. Virol. 2019, 93, 1448–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babayan, S.A.; Orton, R.J.; Streicker, D.G. Predicting reservoir hosts and arthropod vectors from evolutionary signatures in RNA virus genomes. Science 2018, 362, 577–580. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.W.; Dickerman, A.W.; Pineyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.J. Origin, evolution and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. MBio 2013, 4, e00737–e007413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.S.; Knowles, S.; Nashold, S.W.; Ip, H.S.; Leon, A.E.; Rocke, T.; Keller, S.; Carossino, M.; Balasuriya, U.; Hofmeister, E. Experimental challenge of a North American bat species, big brown bat (Eptesicus fuscus), with SARS-CoV-2. Transboud. Emerg. Dis. 2021, 68, 3443–3452. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| County and State | Samples | Positive |
|---|---|---|
| Minnehaha, SD | 112 | 7 |
| Yankton, SD | 6 | 1 |
| Charles Mix, SD | 4 | |
| Turner, SD | 3 | 1 |
| Clay, SD | 1 | |
| Brule, SD | 1 | |
| McCook, SD | 1 | |
| Lake, SD | 5 | |
| Hanson, SD | 1 | |
| Grant, SD | 1 | |
| Brookings, SD | 5 | |
| Codington, SD | 3 | 1 |
| Spink, SD | 1 | |
| Davison, SD | 1 | |
| Hutchinson, SD | 4 | |
| Marshall, SD | 1 | |
| Hughes, SD | 1 | |
| Beadle, SD | 1 | |
| Custer, SD | 2 | |
| Pennington, SD | 2 | |
| Hamlin, SD | 1 | |
| Tripp, SD | 1 | |
| Deuel, SD | 1 | 1 |
| Sanborn, SD | 1 | |
| Hanson, MN | 1 | |
| Lac qui Parle, MN | 1 | |
| Lyon, MN | 4 | |
| Rock, MN | 2 | 1 |
| Redwood, MN | 1 | |
| Pipestone, MN | 1 | |
| Sioux, IA | 2 | |
| Lyon, IA | 1 | |
| Boyd, NE | 1 |
| EbCoV Strain | ORF1ab | S | ORF3 | E | M | N | ORF7 |
|---|---|---|---|---|---|---|---|
| 15,712 | 252–20,845 (6864) | 20,842–24,873 (1343) | 24,870–25,544 (224) | 25,516–25,761 (81) | 25,772–26,449 (225) | 26,467–27,627 (386) | 27,630–28,466 (278) |
| 16,842 | 357–20,356 (6666) * | 20,353–24,384 (1343) | 24,381–25,055 (224) | 25,027–25,272 (81) | 25,283–25,960 (225) | 25,978–27,138 (386) | 27,141–27,977 (278) |
| 16,964 | 97–20,690 (6864) | 20,687–24,730 (1347) | 24,727–25,401 (224) | 25,373–25,618 (81) | 25,629–26,306 (225) | 26,324–27,484 (386) | 27,487–28,323 (278) |
| 14,300 | 366–20,362 (6665) * | 20,359–24,408 (1349) | 24,405–25,079 (224) | 25,051–25,296 (81) | 25,307–25,984 (225) | 26,002–27,159 (385) | 27,162–27,998 (278) |
| 15,073 | 366–20,365 (6666) * | 20,362–24,411 (1349) | 24,408–25,082 (224) | 25,054–25,299 (81) | 25,310–25,987 (225) | 26,005–27,165 (386) | 27,168–28,004 (278) |
| 15,593 | 306–20,905 (6866) | 20,902–24,933 (1343) | 24,930–25,604 (224) | 25,513–25,821 (102) | 25,832–26,509 (225) | 26,527–27,687 (386) | 27,690–28,526 (278) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaeffer, R.; Temeeyasen, G.; Hause, B.M. Alphacoronaviruses Are Common in Bats in the Upper Midwestern United States. Viruses 2022, 14, 184. https://doi.org/10.3390/v14020184
Schaeffer R, Temeeyasen G, Hause BM. Alphacoronaviruses Are Common in Bats in the Upper Midwestern United States. Viruses. 2022; 14(2):184. https://doi.org/10.3390/v14020184
Chicago/Turabian StyleSchaeffer, Reagan, Gun Temeeyasen, and Ben M. Hause. 2022. "Alphacoronaviruses Are Common in Bats in the Upper Midwestern United States" Viruses 14, no. 2: 184. https://doi.org/10.3390/v14020184