
Assessment of Inter-Laboratory Differences in SARS-CoV-2 Consensus Genome Assemblies between Public Health Laboratories in Australia

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. SARS-CoV-2 Matched Samples
2.2. Wet-Lab Protocols
2.3. Bioinformatics Protocols
2.4. Sequence Comparisons
3. Results
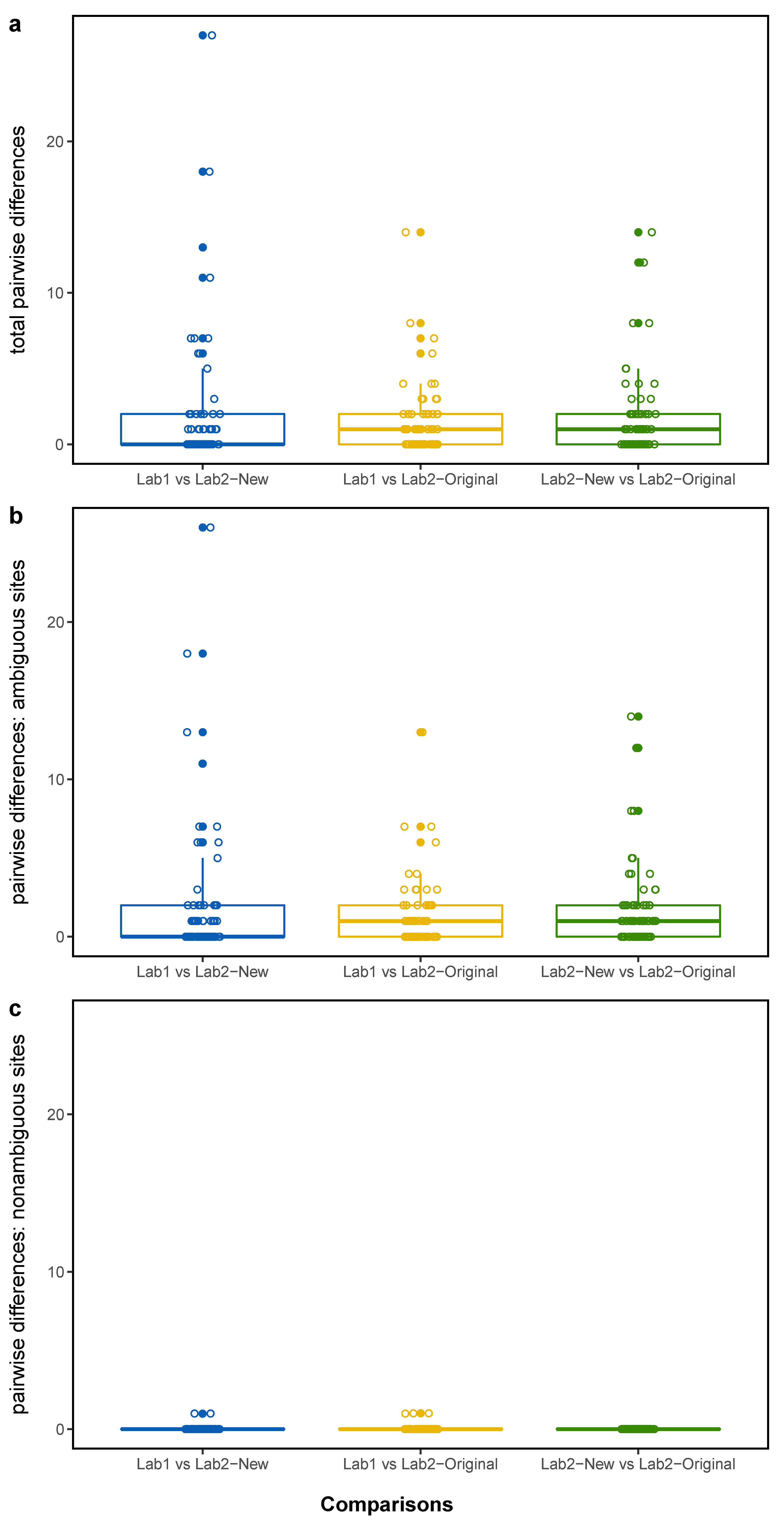
3.1. Pairwise Differences
- Lab1 vs. Lab2-New: Lab1 sample assembled with Lab1 bioinformatics pipeline; Lab2 sample assembled with Lab1 bioinformatics pipeline
- Lab1 vs. Lab2-Original: Lab1 sample assembled with Lab1 bioinformatics pipeline; Lab2 sample assembled with Lab2 bioinformatics pipeline
- Lab2-New vs. Lab2-Original: Lab2 sample assembled with Lab1 bioinformatics pipeline; Lab2 sample assembled with Lab2 bioinformatics pipeline
3.2. Impact on Pango Lineage Assignment
3.3. Impact on Placement in Phylogenetic Tree
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel Coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new Coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Volz, E.M.; Koelle, K.; Bedford, T. Viral Phylodynamics. PLoS Comput. Biol. 2013, 9, e1002947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudas, G.; Carvalho, L.M.; Bedford, T.; Tatem, A.J.; Baele, G.; Faria, N.R.; Park, D.J.; Ladner, J.T.; Arias, A.; Asogun, D.; et al. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 2017, 544, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Alteri, C.; Cento, V.; Piralla, A.; Costabile, V.; Tallarita, M.; Colagrossi, L.; Renica, S.; Giardina, F.; Novazzi, F.; Gaiarsa, S.; et al. genomic epidemiology of SARS-CoV-2 reveals multiple lineages and early spread of SARS-CoV-2 infections in Lombardy, Italy. Nat. Commun. 2021, 12, 434. [Google Scholar] [CrossRef]
- Geoghegan, J.L.; Ren, X.; Storey, M.; Hadfield, J.; Jelley, L.; Jefferies, S.; Sherwood, J.; Paine, S.; Huang, S.; Douglas, J.; et al. Genomic epidemiology reveals transmission patterns and dynamics of SARS-CoV-2 in aotearoa New Zealand. Nat. Commun. 2020, 11, 6351. [Google Scholar] [CrossRef]
- Lu, J.; du Plessis, L.; Liu, Z.; Hill, V.; Kang, M.; Lin, H.; Sun, J.; François, S.; Kraemer, M.U.G.; Faria, N.R.; et al. Genomic epidemiology of SARS-CoV-2 in Guangdong province, China. Cell 2020, 181, 997–1003.e9. [Google Scholar] [CrossRef] [PubMed]
- Rockett, R.J.; Arnott, A.; Lam, C.; Sadsad, R.; Timms, V.; Gray, K.-A.; Eden, J.-S.; Chang, S.; Gall, M.; Draper, J.; et al. Revealing COVID-19 transmission in Australia by SARS-CoV-2 genome sequencing and agent-based modeling. Nat. Med. 2020, 26, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T.; Lane, C.R.; Sherry, N.L.; Duchene, S.; Gonçalves da Silva, A.; Caly, L.; Sait, M.; Ballard, S.A.; Horan, K.; Schultz, M.B.; et al. Tracking the COVID-19 pandemic in Australia using genomics. Nat. Commun. 2020, 11, 4376. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Frampton, D.; Rampling, T.; Cross, A.; Bailey, H.; Heaney, J.; Byott, M.; Scott, R.; Sconza, R.; Price, J.; Margaritis, M.; et al. Genomic characteristics and clinical effect of the emergent SARS-CoV-2 B.1.1.7 lineage in London, UK: A whole-genome sequencing and hospital-based Cohort study. Lancet Infect. Dis. 2021, 21, 1246–1256. [Google Scholar] [CrossRef]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal signal and the Phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull, R.A.; Adikari, T.N.; Ferguson, J.M.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Naing, Z.; Yeang, M.; Verich, A.; Gamaarachchi, H.; et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat. Commun. 2020, 11, 6272. [Google Scholar] [CrossRef]
- De Maio, N.; Walker, C.; Borges, R.; Weilguny, L.; Slodkowicz, G.; Goldman, N. Issues with SARS-CoV-2 Sequencing Data. Available online: https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473 (accessed on 14 April 2021).
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and severity of novel SARS-CoV-2 variant of concern 202012/01 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Walensky, R.P.; Walke, H.T.; Fauci, A.S. SARS-CoV-2 variants of concern in the United States—Challenges and opportunities. JAMA 2021, 325, 1037–1038. [Google Scholar] [CrossRef] [PubMed]
- Zucman, N.; Uhel, F.; Descamps, D.; Roux, D.; Ricard, J.-D. Severe reinfection with South African SARS-CoV-2 variant 501Y.V2: A case report. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2021, 73, 1945–1946. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S. Within-host viral diversity: A window into viral evolution. Annu. Rev. Virol. 2020, 7, 63–81. [Google Scholar] [CrossRef]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef]
- Armero, A.; Berthet, N.; Avarre, J.-C. Intra-host diversity of SARS-CoV-2 should not be neglected: Case of the State of Victoria, Australia. Viruses 2021, 13, 133. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and IVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Deveson, I.W.; Pang, C.N.I.; Yeang, M.; Naing, Z.; Adikari, T.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Verich, A.; et al. Respiratory viral Co-infections among SARS-CoV-2 cases confirmed by virome capture sequencing. Sci. Rep. 2021, 11, 3934. [Google Scholar] [CrossRef]
- Eden, J.-S.; Rockett, R.; Carter, I.; Rahman, H.; de Ligt, J.; Hadfield, J.; Storey, M.; Ren, X.; Tulloch, R.; Basile, K.; et al. An Emergent Clade of SARS-CoV-2 Linked to Returned Travellers from Iran. Virus Evol. 2020, 6, veaa027. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and Sensitive Classification of Metagenomic Sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Andermann, T.; Fernandes, A.M.; Olsson, U.; Töpel, M.; Pfeil, B.; Oxelman, B.; Aleixo, A.; Faircloth, B.C.; Antonelli, A. Allele phasing greatly improves the phylogenetic utility of ultraconserved elements. Syst. Biol. 2019, 68, 32–46. [Google Scholar] [CrossRef]
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.S.; De Maio, N.; Gozashti, L.; Lanfear, R.; Haussler, D.; Corbett-Detig, R. Ultrafast sample placement on existing TRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat. Genet. 2021, 53, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; Maio, N.D.; et al. BEAST 2.5: An advanced software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kates, H.R.; Johnson, M.G.; Gardner, E.M.; Zerega, N.J.C.; Wickett, N.J. Allele phasing has minimal impact on phylogenetic reconstruction from targeted nuclear gene sequences in a case study of artocarpus. Am. J. Bot. 2018, 105, 404–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| IUPAC Ambiguities Included | IUPAC Ambiguities Excluded | |||||||
|---|---|---|---|---|---|---|---|---|
| Pair | Mean | Median | IQR | Range | Mean | Median | IQR | Range |
| Lab1 vs. Lab2-New | 1.97 | 0 | 2 | 0–27 | 0.03 | 0 | 0 | 0–1 |
| Lab1 vs. Lab2-Original | 1.32 | 1 | 2 | 0–14 | 0.04 | 0 | 0 | 0–1 |
| Lab2-New vs. Lab2-Original | 1.72 | 1 | 2 | 0–14 | 0 | 0 | 0 | 0–0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foster, C.S.P.; Stelzer-Braid, S.; Deveson, I.W.; Bull, R.A.; Yeang, M.; Au, J.-P.; Ruiz Silva, M.; van Hal, S.J.; Rockett, R.J.; Sintchenko, V.; et al. Assessment of Inter-Laboratory Differences in SARS-CoV-2 Consensus Genome Assemblies between Public Health Laboratories in Australia. Viruses 2022, 14, 185. https://doi.org/10.3390/v14020185
Foster CSP, Stelzer-Braid S, Deveson IW, Bull RA, Yeang M, Au J-P, Ruiz Silva M, van Hal SJ, Rockett RJ, Sintchenko V, et al. Assessment of Inter-Laboratory Differences in SARS-CoV-2 Consensus Genome Assemblies between Public Health Laboratories in Australia. Viruses. 2022; 14(2):185. https://doi.org/10.3390/v14020185
Chicago/Turabian StyleFoster, Charles S. P., Sacha Stelzer-Braid, Ira W. Deveson, Rowena A. Bull, Malinna Yeang, Jane-Phan Au, Mariana Ruiz Silva, Sebastiaan J. van Hal, Rebecca J. Rockett, Vitali Sintchenko, and et al. 2022. "Assessment of Inter-Laboratory Differences in SARS-CoV-2 Consensus Genome Assemblies between Public Health Laboratories in Australia" Viruses 14, no. 2: 185. https://doi.org/10.3390/v14020185