
U5 snRNP Core Proteins Are Key Components of the Defense Response against Viral Infection through Their Roles in Programmed Cell Death and Interferon Induction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Viruses, and Treatments
2.2. Viral Infection
2.3. siRNA Transfection
2.4. Real-Time Cell Analysis (RTCA)
2.5. FACS Analysis
2.6. Methylene Blue Staining
2.7. High-Throughput Multiplex Microscopy-Based Apoptosis Assay
2.8. Caspase Activity Assay
2.9. RNA Extraction
2.10. Reverse Transcription
2.11. qPCR
2.12. Alternative Splicing PCR (AS-PCR)
2.13. Immunoblotting Analysis
2.14. Infectious Viral Titers Using TCID50
2.15. Data Analysis and Statistical Analyses
2.16. Data Availability
3. Results
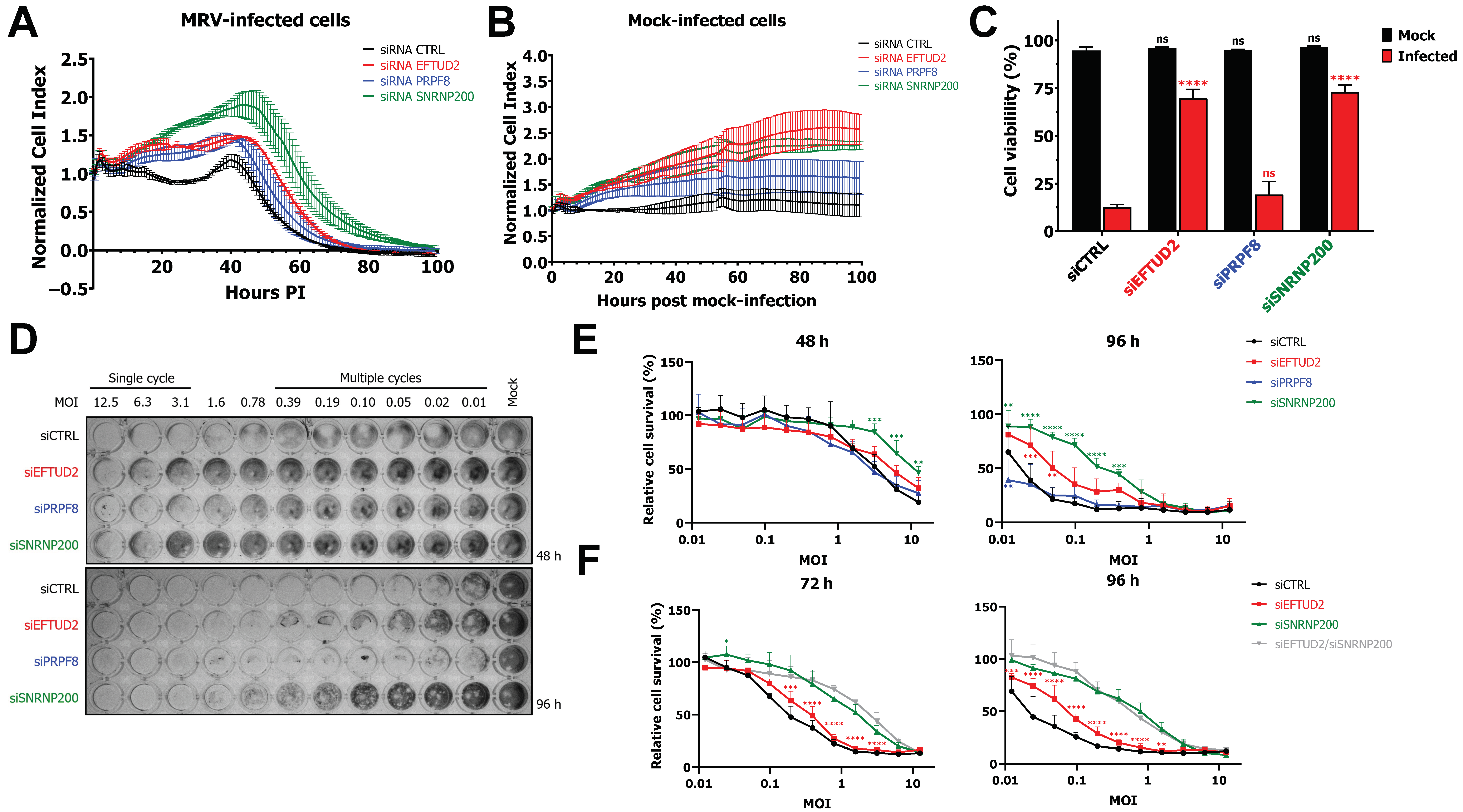
3.1. EFTUD2 and SNRNP200 Are Required for Cell Death after Infection
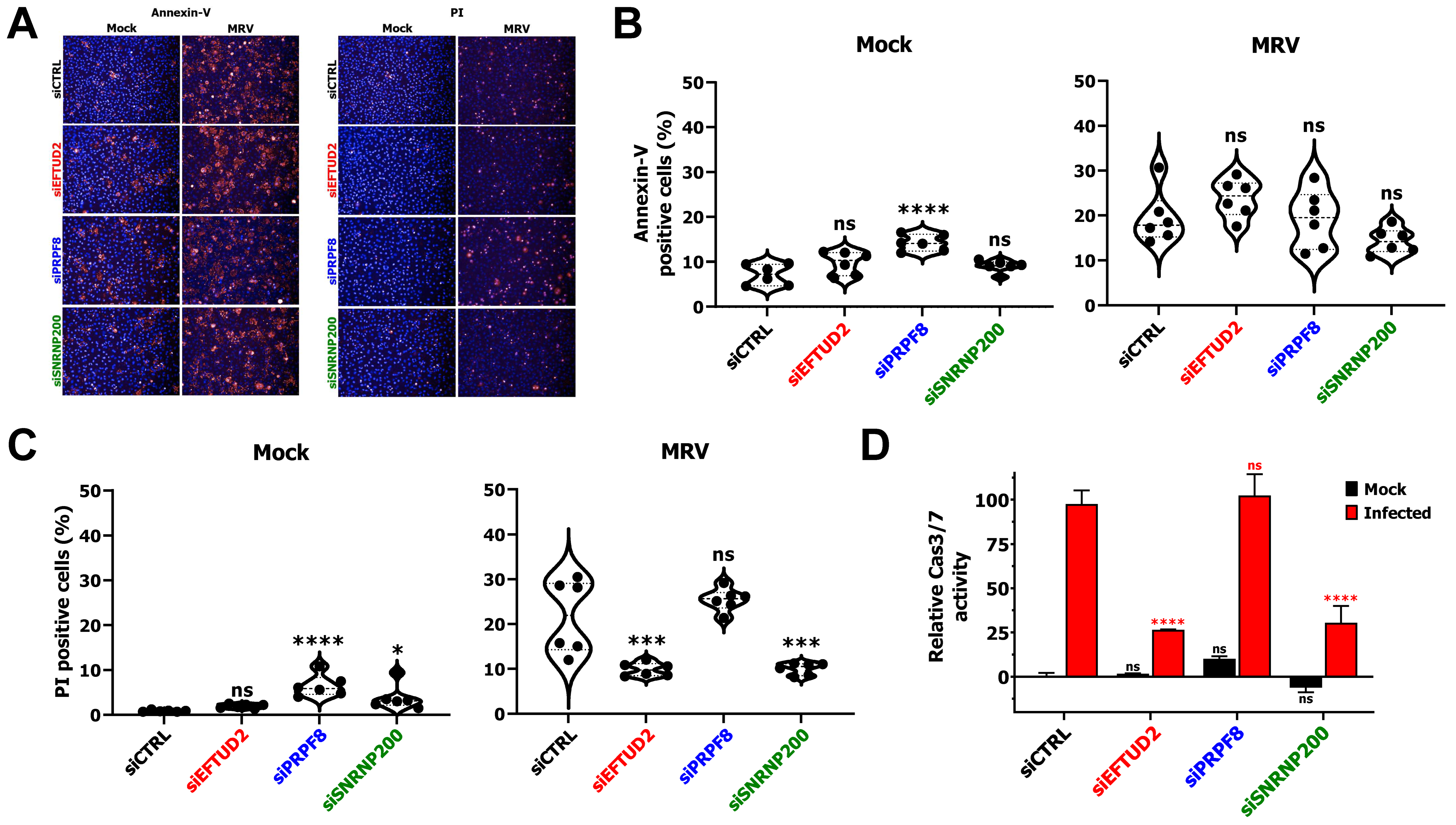
3.2. EFTUD2 and SNRNP200 Knock-Down Affects Apoptosis
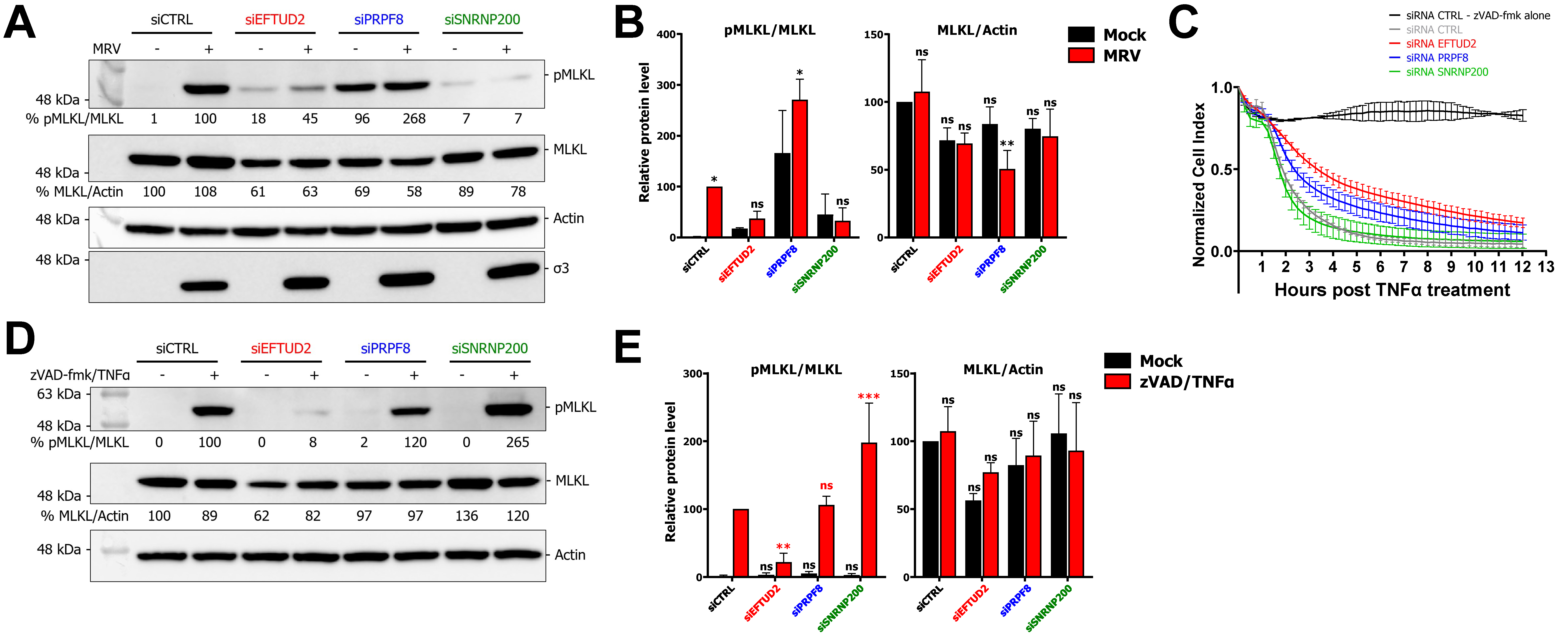
3.3. EFTUD2 and SNRNP200 Are Required for Necroptosis
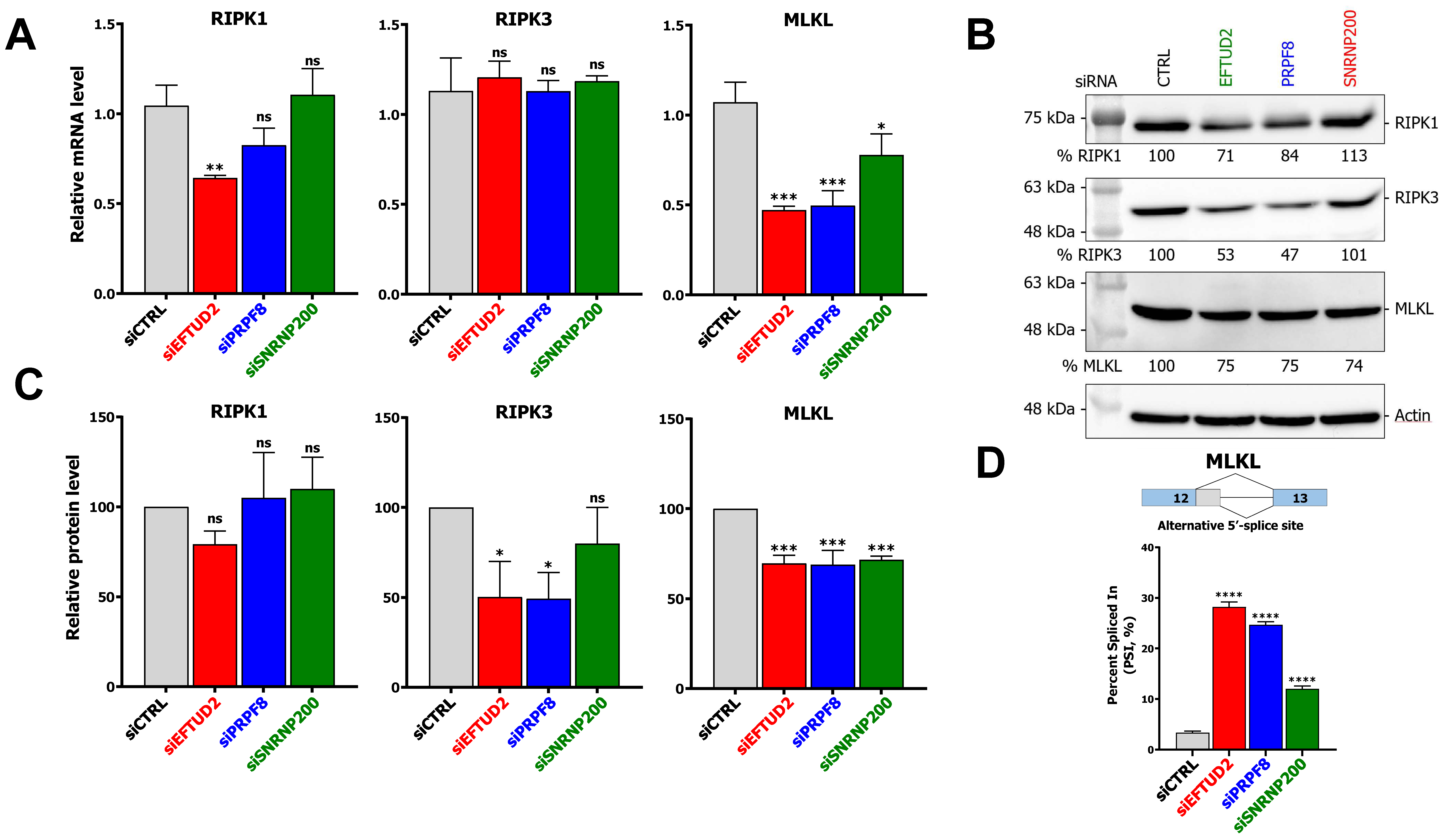
3.4. KD of U5 snRNP Core Components Impacts mRNA Levels, Protein Levels, and Alternative Splicing of Necroptotic Regulators
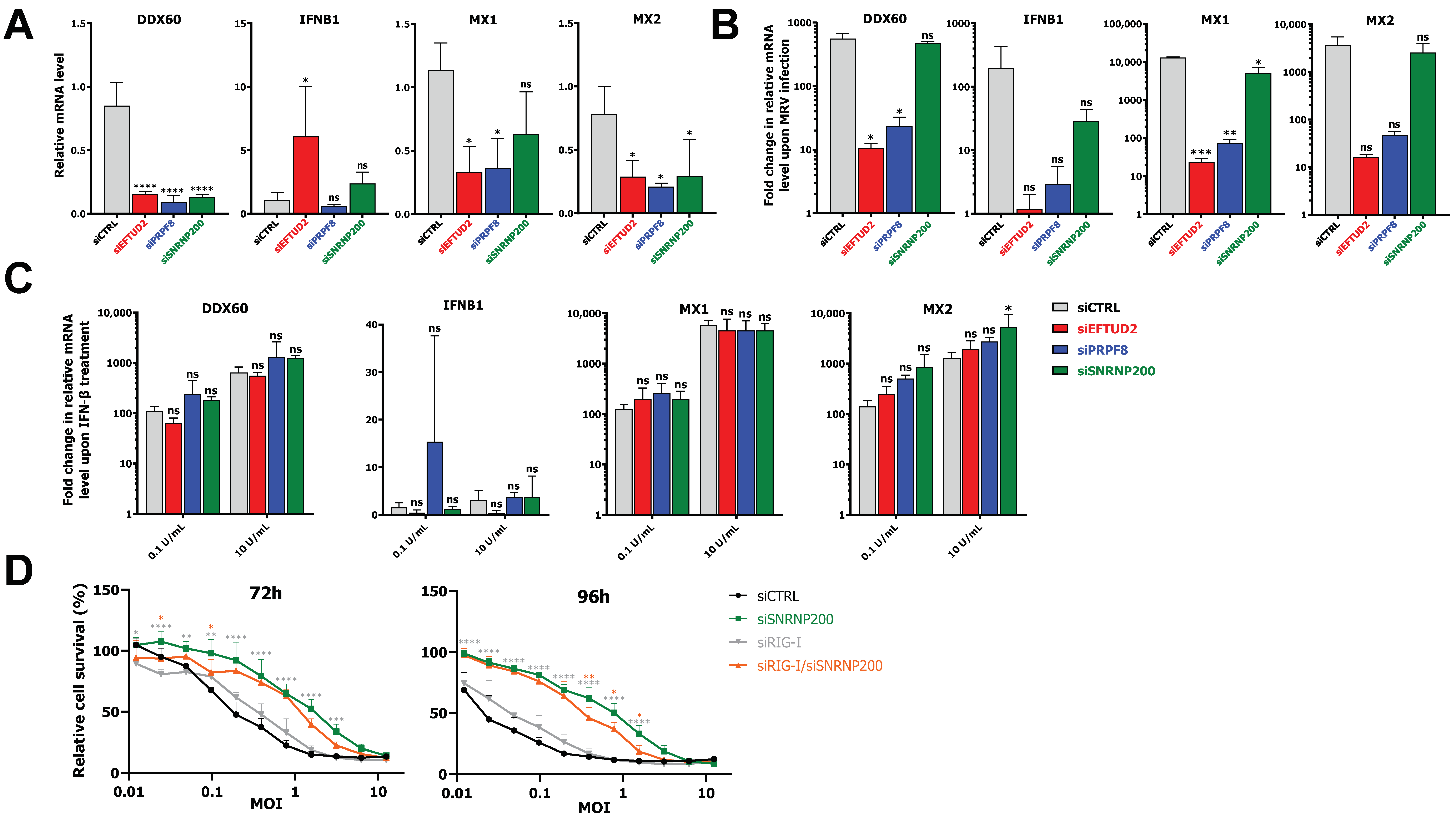
3.5. EFTUD2 and PRPF8 Are Required for Optimal Induction of the IFN Response
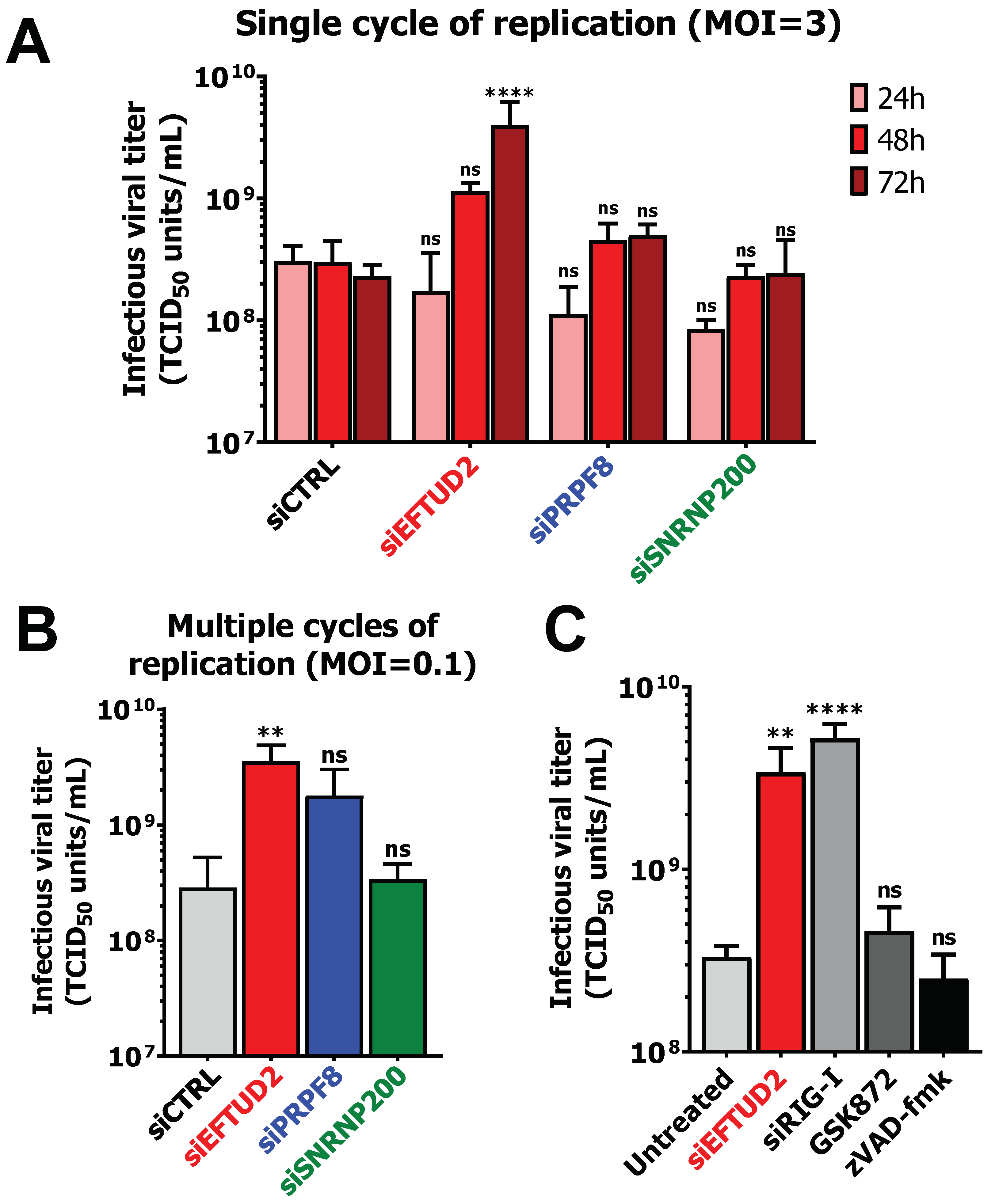
3.6. EFTUD2 Restricts MRV’s Replication in Both Single Cycle and Multiple Cycles of Replication
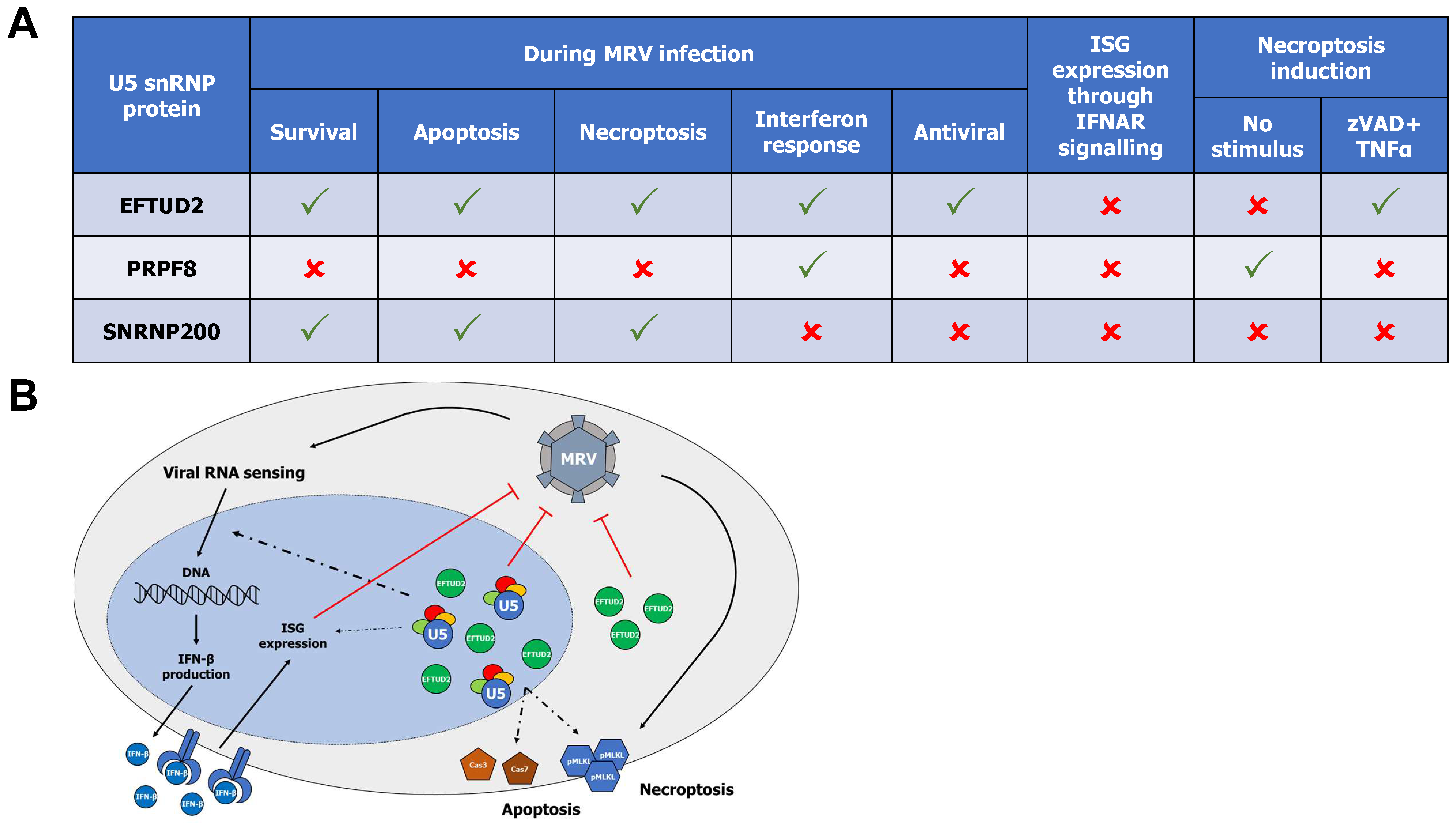
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fensterl, V.; Chattopadhyay, S.; Sen, G.C. No Love Lost Between Viruses and Interferons. Annu. Rev. Virol. 2015, 2, 549–572. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.C.; Sarkar, S.N. The Interferon-Stimulated Genes: Targets of Direct Signaling by Interferons, Double-Stranded RNA, and Viruses. Curr. Top. Microbiol. Immunol. 2007, 316, 233–250. [Google Scholar] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Maio, F.A.D.; Risso, G.; Iglesias, N.G.; Shah, P.; Pozzi, B.; Gebhard, L.G.; Mammi, P.; Mancini, E.; Yanovsky, M.J.; Andino, R.; et al. The Dengue Virus NS5 Protein Intrudes in the Cellular Spliceosome and Modulates Splicing. PLoS Pathog. 2016, 12, e1005841. [Google Scholar] [CrossRef] [Green Version]
- Boudreault, S.; Durand, M.; Martineau, C.-A.; Perreault, J.-P.; Lemay, G.; Bisaillon, M. Reovirus M2 Protein Modulates Host Cell Alternative Splicing by Reducing Protein Levels of U5 SnRNP Core Components. Nucleic Acids Res. 2022, 50, 5263–5281. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-C.; Kuo, R.-L.; Lin, J.-Y.; Huang, P.-N.; Huang, Y.; Liu, H.; Arnold, J.J.; Chen, S.-J.; Wang, R.Y.-L.; Cameron, C.E.; et al. Cytoplasmic Viral RNA-Dependent RNA Polymerase Disrupts the Intracellular Splicing Machinery by Entering the Nucleus and Interfering with Prp8. PLoS Pathog. 2014, 10, e1004199. [Google Scholar] [CrossRef] [PubMed]
- Boudreault, S.; Roy, P.; Lemay, G.; Bisaillon, M. Viral Modulation of Cellular RNA Alternative Splicing: A New Key Player in Virus–Host Interactions? Wiley Interdiscip. Rev. RNA 2019, 10, e1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Rauhut, R.; Vornlocher, H.-P.; Lührmann, R. The Network of Protein–Protein Interactions within the Human U4/U6.U5 Tri-SnRNP. RNA 2006, 12, 1418–1430. [Google Scholar] [CrossRef] [Green Version]
- Wood, K.A.; Eadsforth, M.A.; Newman, W.G.; O’Keefe, R.T. The Role of the U5 SnRNP in Genetic Disorders and Cancer. Front. Genet 2021, 12, 636620. [Google Scholar] [CrossRef]
- Newman, A.J. The Role of U5 SnRNP in Pre-MRNA Splicing. EMBO J. 1997, 16, 5797–5800. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, D.L. Mechanisms of Alternative Pre-Messenger RNA Splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-MRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Goren, A.; Ast, G. Alternative Splicing: Current Perspectives. BioEssays 2008, 30, 38–47. [Google Scholar] [CrossRef]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative Splicing and Evolution: Diversification, Exon Definition and Function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Xiao, R.; Blue, S.M.; Chen, J.-Y.; Cody, N.A.L.; Dominguez, D.; et al. A Large-Scale Binding and Functional Map of Human RNA-Binding Proteins. Nature 2020, 583, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, K.; Kalam, H.; Dutt, R.; Kumar, D. RNA Splicing: A New Paradigm in Host-Pathogen Interactions. J. Mol. Biol. 2019, 431, 1565–1575. [Google Scholar] [CrossRef]
- Ashraf, U.; Benoit-Pilven, C.; Lacroix, V.; Navratil, V.; Naffakh, N. Advances in Analyzing Virus-Induced Alterations of Host Cell Splicing. Trends Microbiol. 2019, 27, 268–281. [Google Scholar] [CrossRef]
- Cross, S.T.; Michalski, D.; Miller, M.R.; Wilusz, J. RNA Regulatory Processes in RNA Virus Biology. Wiley Interdiscip. Rev. RNA 2019, 10, e1536. [Google Scholar] [CrossRef] [Green Version]
- Lemay, G. Synthesis and Translation of Viral MRNA in Reovirus-Infected Cells: Progress and Remaining Questions. Viruses 2018, 10, 671. [Google Scholar] [CrossRef]
- Danthi, P.; Guglielmi, K.M.; Kirchner, E.; Mainou, B.; Stehle, T.; Dermody, T.S. From Touchdown to Transcription: The Reovirus Cell Entry Pathway. Curr Top Microbiol Immunol 2010, 343, 91–119. [Google Scholar] [CrossRef] [PubMed]
- Danthi, P.; Holm, G.H.; Stehle, T.; Dermody, T.S. Reovirus Receptors, Cell Entry, and Proapoptotic Signaling. Adv. Exp. Med. Biol. 2013, 790, 42–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreault, S.; Martenon-Brodeur, C.; Caron, M.; Garant, J.-M.; Tremblay, M.-P.; Armero, V.E.S.; Durand, M.; Lapointe, E.; Thibault, P.; Tremblay-Létourneau, M.; et al. Global Profiling of the Cellular Alternative RNA Splicing Landscape during Virus-Host Interactions. PLoS ONE 2016, 11, e0161914. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Serrano, E.E.; Fritch, E.J.; Scholl, E.H.; Sherry, B. A Cytoplasmic RNA Virus Alters the Function of the Cell Splicing Protein SRSF2. J. Virol. 2017, 91, e02488-16. [Google Scholar] [CrossRef] [Green Version]
- Danis, C.; Lemay, G. Protein Synthesis in Different Cell Lines Infected with Orthoreovirus Serotype 3: Inhibition of Host-Cell Protein Synthesis Correlates with Accelerated Viral Multiplication and Cell Killing. Biochem. Cell Biol. 1993, 71, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Lanoie, D.; Lemay, G. Multiple Proteins Differing between Laboratory Stocks of Mammalian Orthoreoviruses Affect Both Virus Sensitivity to Interferon and Induction of Interferon Production during Infection. Virus Res. 2018, 247, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Sandekian, V.; Lemay, G. A Single Amino Acid Substitution in the MRNA Capping Enzyme Λ2 of a Mammalian Orthoreovirus Mutant Increases Interferon Sensitivity. Virology 2015, 483, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Antar, A.A.R.; Boehme, K.W.; Danthi, P.; Eby, E.A.; Guglielmi, K.M.; Holm, G.H.; Johnson, E.M.; Maginnis, M.S.; Naik, S.; et al. A Plasmid-Based Reverse Genetics System for Animal Double-Stranded RNA Viruses. Cell Host Microbe 2007, 1, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Prinos, P.; Garneau, D.; Lucier, J.-F.; Gendron, D.; Couture, S.; Boivin, M.; Brosseau, J.-P.; Lapointe, E.; Thibault, P.; Durand, M.; et al. Alternative Splicing of SYK Regulates Mitosis and Cell Survival. Nat. Struct. Mol. Biol. 2011, 18, 673–679. [Google Scholar] [CrossRef]
- Faucher-Giguère, L.; Roy, A.; Deschamps-Francoeur, G.; Couture, S.; Nottingham, R.M.; Lambowitz, A.M.; Scott, M.S.; Abou Elela, S. High-Grade Ovarian Cancer Associated H/ACA SnoRNAs Promote Cancer Cell Proliferation and Survival. NAR Cancer 2022, 4, zcab050. [Google Scholar] [CrossRef]
- Taylor, S.; Wakem, M.; Dijkman, G.; Alsarraj, M.; Nguyen, M. A Practical Approach to RT-QPCR—Publishing Data That Conform to the MIQE Guidelines. Methods 2010, 50, S1–S5. [Google Scholar] [CrossRef]
- Virgin, H.W.; Mann, M.A.; Fields, B.N.; Tyler, K.L. Monoclonal Antibodies to Reovirus Reveal Structure/Function Relationships between Capsid Proteins and Genetics of Susceptibility to Antibody Action. J. Virol. 1991, 65, 6772–6781. [Google Scholar] [CrossRef] [Green Version]
- Blanca, M.J.; Alarcón, R.; Arnau, J. Non-Normal Data: Is ANOVA Still a Valid Option? Psicothema 2017, 29, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Lebourgeois, S.; Fraisse, A.; Hennechart-Collette, C.; Guillier, L.; Perelle, S.; Martin-Latil, S. Development of a Real-Time Cell Analysis (RTCA) Method as a Fast and Accurate Method for Detecting Infectious Particles of the Adapted Strain of Hepatitis A Virus. Front. Cell. Infect. Microbiol. 2018, 8, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burmakina, G.; Bliznetsov, K.; Malogolovkin, A. Real-Time Analysis of the Cytopathic Effect of African Swine Fever Virus. J. Virol. Methods 2018, 257, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Ye, P.; Wang, X.; Xu, X.; Reisen, W. Real-Time Monitoring of Flavivirus Induced Cytopathogenesis Using Cell Electric Impedance Technology. J. Virol. Methods 2011, 173, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charretier, C.; Saulnier, A.; Benair, L.; Armanet, C.; Bassard, I.; Daulon, S.; Bernigaud, B.; Rodrigues de Sousa, E.; Gonthier, C.; Zorn, E.; et al. Robust Real-Time Cell Analysis Method for Determining Viral Infectious Titers during Development of a Viral Vaccine Production Process. J. Virol. Methods 2018, 252, 57–64. [Google Scholar] [CrossRef]
- Teng, Z.; Kuang, X.; Wang, J.; Zhang, X. Real-Time Cell Analysis—A New Method for Dynamic, Quantitative Measurement of Infectious Viruses and Antiserum Neutralizing Activity. J. Virol. Methods 2013, 193, 364–370. [Google Scholar] [CrossRef] [Green Version]
- Lanoie, D.; Côté, S.; Degeorges, E.; Lemay, G. A Single Mutation in the Mammalian Orthoreovirus S1 Gene Is Responsible for Increased Interferon Sensitivity in a Virus Mutant Selected in Vero Cells. Virology 2019, 528, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Sandekian, V.; Lim, D.; Prud’homme, P.; Lemay, G. Transient High Level Mammalian Reovirus Replication in a Bat Epithelial Cell Line Occurs without Cytopathic Effect. Virus Res. 2013, 173, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Holm, G.H.; Zurney, J.; Tumilasci, V.; Leveille, S.; Danthi, P.; Hiscott, J.; Sherry, B.; Dermody, T.S. Retinoic Acid-Inducible Gene-I and Interferon-β Promoter Stimulator-1 Augment Proapoptotic Responses Following Mammalian Reovirus Infection via Interferon Regulatory Factor-3. J. Biol. Chem. 2007, 282, 21953–21961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyler, K.L.; Squier, M.K.; Rodgers, S.E.; Schneider, B.E.; Oberhaus, S.M.; Grdina, T.A.; Cohen, J.J.; Dermody, T.S. Differences in the Capacity of Reovirus Strains to Induce Apoptosis Are Determined by the Viral Attachment Protein Sigma 1. J. Virol. 1995, 69, 6972–6979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, P.; DeBiasi, R.L.; Meintzer, S.M.; Robinson, B.A.; Tyler, K.L. Inhibition of NF-κ B Activity and CFLIP Expression Contribute to Viral-Induced Apoptosis. Apoptosis 2005, 10, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.K.; Hiller, B.E.; Thete, D.; Snyder, A.J.; Perez, E.; Upton, J.W.; Danthi, P. Viral RNA at Two Stages of Reovirus Infection Is Required for the Induction of Necroptosis. J. Virol. 2017, 91, e02404-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.K.; Danthi, P. Reovirus Activates a Caspase-Independent Cell Death Pathway. mBio 2013, 4, e00178-13. [Google Scholar] [CrossRef] [Green Version]
- Nagata, S.; Suzuki, J.; Segawa, K.; Fujii, T. Exposure of Phosphatidylserine on the Cell Surface. Cell Death Differ 2016, 23, 952–961. [Google Scholar] [CrossRef] [Green Version]
- Shlomovitz, I.; Speir, M.; Gerlic, M. Flipping the Dogma—Phosphatidylserine in Non-Apoptotic Cell Death. Cell Commun. Signal. 2019, 17, 139. [Google Scholar] [CrossRef] [Green Version]
- Koehler, H.; Cotsmire, S.; Langland, J.; Kibler, K.V.; Kalman, D.; Upton, J.W.; Mocarski, E.S.; Jacobs, B.L. Inhibition of DAI-Dependent Necroptosis by the Z-DNA Binding Domain of the Vaccinia Virus Innate Immune Evasion Protein, E3. Proc. Natl. Acad. Sci. USA 2017, 114, 11506–11511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehler, H.; Cotsmire, S.; Zhang, T.; Balachandran, S.; Upton, J.W.; Langland, J.; Kalman, D.; Jacobs, B.L.; Mocarski, E.S. Vaccinia Virus E3 Prevents Sensing of Z-RNA to Block ZBP1-Dependent Necroptosis. Cell Host Microbe 2021, 29, 1266–1276.e5. [Google Scholar] [CrossRef]
- Patzina, C.; Botting, C.H.; García-Sastre, A.; Randall, R.E.; Hale, B.G. Human Interactome of the Influenza B Virus NS1 Protein. J. Gen. Virol. 2017, 98, 2267–2273. [Google Scholar] [CrossRef]
- Tremblay, N.; Baril, M.; Chatel-Chaix, L.; Es-Saad, S.; Park, A.Y.; Koenekoop, R.K.; Lamarre, D. Spliceosome SNRNP200 Promotes Viral RNA Sensing and IRF3 Activation of Antiviral Response. PLoS Pathog. 2016, 12, e1005772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Xiao, F.; Hong, J.; Wang, K.; Liu, X.; Cai, D.; Fusco, D.N.; Zhao, L.; Jeong, S.W.; Brisac, C.; et al. EFTUD2 Is a Novel Innate Immune Regulator Restricting Hepatitis C Virus Infection through the RIG-I/MDA5 Pathway. J. Virol. 2015, 89, 6608–6618. [Google Scholar] [CrossRef] [Green Version]
- De Arras, L.; Laws, R.; Leach, S.M.; Pontis, K.; Freedman, J.H.; Schwartz, D.A.; Alper, S. Comparative Genomics RNAi Screen Identifies Eftud2 as a Novel Regulator of Innate Immunity. Genetics 2014, 197, 485–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schock, S.N.; Chandra, N.V.; Sun, Y.; Irie, T.; Kitagawa, Y.; Gotoh, B.; Coscoy, L.; Winoto, A. Induction of Necroptotic Cell Death by Viral Activation of the RIG-I or STING Pathway. Cell Death Differ. 2017, 24, 615–625. [Google Scholar] [CrossRef]
- Mohamed, A.; Konda, P.; Eaton, H.E.; Gujar, S.; Smiley, J.R.; Shmulevitz, M. Closely Related Reovirus Lab Strains Induce Opposite Expression of RIG-I/IFN-Dependent versus -Independent Host Genes, via Mechanisms of Slow Replication versus Polymorphisms in DsRNA Binding Σ3 Respectively. PLoS Pathog. 2020, 16, e1008803. [Google Scholar] [CrossRef]
- Thoresen, D.; Wang, W.; Galls, D.; Guo, R.; Xu, L.; Pyle, A.M. The Molecular Mechanism of RIG-I Activation and Signaling. Immunol. Rev. 2021, 304, 154–168. [Google Scholar] [CrossRef]
- Zarbl, H.; Millward, S. The Reovirus Multiplication Cycle. In The Reoviridae; Joklik, W.K., Ed.; The Viruses; Springer US: Boston, MA, USA, 1983; pp. 107–196. ISBN 978-1-4899-0580-2. [Google Scholar]
- Santiana, M.; Ghosh, S.; Ho, B.A.; Rajasekaran, V.; Du, W.-L.; Mutsafi, Y.; Jésus-Diaz, D.A.D.; Sosnovtsev, S.V.; Levenson, E.A.; Parra, G.I.; et al. Vesicle-Cloaked Virus Clusters Are Optimal Units for Inter-Organismal Viral Transmission. Cell Host Microbe 2018, 24, 208–220.e8. [Google Scholar] [CrossRef] [Green Version]
- Fernández de Castro, I.; Tenorio, R.; Ortega-González, P.; Knowlton, J.J.; Zamora, P.F.; Lee, C.H.; Fernández, J.J.; Dermody, T.S.; Risco, C. A Modified Lysosomal Organelle Mediates Nonlytic Egress of Reovirus. J. Cell Biol. 2020, 219, e201910131. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.M.; Mainou, B.A.; Kim, K.S.; Dermody, T.S. Directional Release of Reovirus from the Apical Surface of Polarized Endothelial Cells. mBio 2013, 4, e00049-13. [Google Scholar] [CrossRef] [Green Version]
- Stanković, D.; Claudius, A.-K.; Schertel, T.; Bresser, T.; Uhlirova, M. A Drosophila Model to Study Retinitis Pigmentosa Pathology Associated with Mutations in the Core Splicing Factor Prp8. Dis. Model. Mech. 2020, 13, dmm043174. [Google Scholar] [CrossRef]
- Xu, Q.; Deng, B.; Li, M.; Chen, Y.; Zhuan, L. CircRNA-UBAP2 Promotes the Proliferation and Inhibits Apoptosis of Ovarian Cancer Though MiR-382-5p/PRPF8 Axis. J. Ovarian Res. 2020, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.; He, L.; You, Y.; Li, J.; Yao, N.; Qu, C.; Huang, W.; Xu, L.; Luo, R.; Hong, J. EFTUD2 Maintains the Survival of Tumor Cells and Promotes Hepatocellular Carcinoma Progression via the Activation of STAT3. Cell Death Dis 2020, 11, 830. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Yan, S.-Y.; Yang, R.; Chen, J.-Y.; Li, Y.; Bu, Y.; Chang, N.; Zhou, Q.; Zhu, X.; Li, C.-Y.; et al. Spliceosomal Protein Eftud2 Mutation Leads to P53-Dependent Apoptosis in Zebrafish Neural Progenitors. Nucleic Acids Res. 2017, 45, 3422–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, N.; Maeda, M.; Sugiyama, M.; Ito, S.; Hyodo, T.; Masuda, A.; Tsunoda, N.; Kokuryo, T.; Hamaguchi, M.; Nagino, M.; et al. Inhibition of SNW1 Association with Spliceosomal Proteins Promotes Apoptosis in Breast Cancer Cells. Cancer Med. 2015, 4, 268–277. [Google Scholar] [CrossRef]
- Wood, K.A.; Rowlands, C.F.; Qureshi, W.M.S.; Thomas, H.B.; Buczek, W.A.; Briggs, T.A.; Hubbard, S.J.; Hentges, K.E.; Newman, W.G.; O’Keefe, R.T. Disease Modeling of Core Pre-MRNA Splicing Factor Haploinsufficiency. Hum. Mol. Genet. 2019, 28, 3704–3723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129.e13. [Google Scholar] [CrossRef]
- Yoon, S.; Bogdanov, K.; Kovalenko, A.; Wallach, D. Necroptosis Is Preceded by Nuclear Translocation of the Signaling Proteins That Induce It. Cell Death Differ 2016, 23, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.; Roelandt, R.; Bruggeman, I.; Estornes, Y.; Vandenabeele, P. Nuclear RIPK3 and MLKL Contribute to Cytosolic Necrosome Formation and Necroptosis. Commun Biol 2018, 1, 6. [Google Scholar] [CrossRef] [Green Version]
- Desroches, A.; Denault, J.-B. Caspase-7 Uses RNA to Enhance Proteolysis of Poly(ADP-Ribose) Polymerase 1 and Other RNA-Binding Proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 21521–21528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desroches, A.; Denault, J.-B. Characterization of Caspase-7 Interaction with RNA. Biochem. J. 2021, 478, 2681–2696. [Google Scholar] [CrossRef]
- Mukhopadhyay, U.; Patra, U.; Chandra, P.; Saha, P.; Gope, A.; Dutta, M.; Chawla-Sarkar, M. Rotavirus Activates MLKL-Mediated Host Cellular Necroptosis Concomitantly with Apoptosis to Facilitate Dissemination of Viral Progeny. Mol. Microbiol. 2022, 117, 818–836. [Google Scholar] [CrossRef] [PubMed]
- Soliman, M.; Seo, J.-Y.; Baek, Y.-B.; Park, J.-G.; Kang, M.-I.; Cho, K.-O.; Park, S.-I. Opposite Effects of Apoptotic and Necroptotic Cellular Pathways on Rotavirus Replication. J. Virol. 2022, 96, e01222-21. [Google Scholar] [CrossRef] [PubMed]
- DeAntoneo, C.; Danthi, P.; Balachandran, S. Reovirus Activated Cell Death Pathways. Cells 2022, 11, 1757. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.W.; Hammer, K.; Tollefson, W.C.; Konopka-Anstadt, J.L.; Kobayashi, T.; Dermody, T.S. Nonstructural Protein Σ1s Mediates Reovirus-Induced Cell Cycle Arrest and Apoptosis. J. Virol. 2013, 87, 12967–12979. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of Double-Stranded RNA and Activation of NF-ΚB by Toll-like Receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Abad, A.T.; Danthi, P. Early Events in Reovirus Infection Influence Induction of Innate Immune Response. J. Virol. 2022, 96, e00917-22. [Google Scholar] [CrossRef]
- An, Y.; Wang, X.; Wu, X.; Chen, L.; Yang, Y.; Lin, X.; Wang, N.; Duan, J.; Long, S.; Zhao, X. Oncolytic Reovirus Induces Ovarian Cancer Cell Apoptosis in a TLR3-Dependent Manner. Virus Res. 2021, 301, 198440. [Google Scholar] [CrossRef]
- Katayama, Y.; Tachibana, M.; Kurisu, N.; Oya, Y.; Terasawa, Y.; Goda, H.; Kobiyama, K.; Ishii, K.J.; Akira, S.; Mizuguchi, H.; et al. Oncolytic Reovirus Inhibits Immunosuppressive Activity of Myeloid-Derived Suppressor Cells in a TLR3-Dependent Manner. J. Immunol. 2018, 200, 2987–2999. [Google Scholar] [CrossRef] [Green Version]
- Long, S.; Gu, Y.; An, Y.; Lin, X.; Chen, X.; Wang, X.; Liao, C.; Ouyang, W.; Wang, N.; He, Z.; et al. Reovirus Enhances Cytotoxicity of Natural Killer Cells against Colorectal Cancer via TLR3 Pathway. J. Transl. Med. 2021, 19, 185. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, K.H.; Richardson-Burns, S.; Alexopoulou, L.; Tyler, K.L.; Flavell, R.A.; Oldstone, M.B.A. Does Toll-like Receptor 3 Play a Biological Role in Virus Infections? Virology 2004, 322, 231–238. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 10, gkac194. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boudreault, S.; Lemay, G.; Bisaillon, M. U5 snRNP Core Proteins Are Key Components of the Defense Response against Viral Infection through Their Roles in Programmed Cell Death and Interferon Induction. Viruses 2022, 14, 2710. https://doi.org/10.3390/v14122710
Boudreault S, Lemay G, Bisaillon M. U5 snRNP Core Proteins Are Key Components of the Defense Response against Viral Infection through Their Roles in Programmed Cell Death and Interferon Induction. Viruses. 2022; 14(12):2710. https://doi.org/10.3390/v14122710
Chicago/Turabian StyleBoudreault, Simon, Guy Lemay, and Martin Bisaillon. 2022. "U5 snRNP Core Proteins Are Key Components of the Defense Response against Viral Infection through Their Roles in Programmed Cell Death and Interferon Induction" Viruses 14, no. 12: 2710. https://doi.org/10.3390/v14122710