
Endoplasmic Reticulum Stress in Hepatitis B Virus and Hepatitis C Virus Infection
 , ,
, ,
Abstract
:1. Introduction
2. ER Stress in the Liver
2.1. Risk Factors for ER Stress
2.2. The ER Stress Response and Related Mechanisms
2.2.1. UPR in the Liver
2.2.2. Other ER Stress Response Pathways in the Liver
3. Viruses and the ER Stress Response
3.1. Viruses and the IRE1α Arm of the UPR
3.2. Viruses and the PERK Arm of the UPR
3.3. Viruses and the ATF6 Arm of the UPR
4. HBV and ER Stress
4.1. HBsAg and ER Stress
4.2. HBx and ER Stress
4.3. HBcAg and ER Stress
4.4. Other Factors Related to HBV and ER Stress
5. HCV and ER Stress
5.1. HCV Structural Proteins and ER Stress
5.2. HCV-Related Non-Structural Proteins and ER Stress
5.3. HCV-Related Lipid Metabolism Changes and ER Stress
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization (WHO). Global Hepatitis Report 2017. Available online: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 26 September 2022).
- Lanini, S.; Ustianowski, A.; Pisapia, R.; Zumla, A.; Ippolito, G. Viral Hepatitis: Etiology, Epidemiology, Transmission, Diagnostics, Treatment, and Prevention. Infect. Dis. Clin. N. Am. 2019, 33, 1045–1062. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Friedman, S.L.; Goossens, N.; Hoshida, Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J. Hepatol. 2018, 68, 526–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, S.; Devincenzo, J.P.; Toovey, S.; Wu, J.Z.; Whitley, R.J. Comparison of antiviral resistance across acute and chronic viral infections. Antivir. Res. 2018, 158, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. CMLS 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Shen, Y.; Song, Y.; Zhang, Y.; Zhang, C.; Ma, Y.; Zhang, F.; Chen, L. ER stress-related molecules induced by Hantaan virus infection in differentiated THP-1 cells. Cell Stress Chaperones 2021, 26, 41–50. [Google Scholar] [CrossRef]
- Xia, S.W.; Wang, Z.M.; Sun, S.M.; Su, Y.; Li, Z.H.; Shao, J.J.; Tan, S.Z.; Chen, A.P.; Wang, S.J.; Zhang, Z.L.; et al. Endoplasmic reticulum stress and protein degradation in chronic liver disease. Pharmacol. Res. 2020, 161, 105218. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kyaw, Y.Y.; Cheong, J. Functional interaction of endoplasmic reticulum stress and hepatitis B virus in the pathogenesis of liver diseases. World J. Gastroenterol. 2017, 23, 7657–7665. [Google Scholar] [CrossRef]
- Kaplowitz, N.; Than, T.A.; Shinohara, M.; Ji, C. Endoplasmic reticulum stress and liver injury. Semin. Liver Dis. 2007, 27, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.K.; Cheong, K.J.; Kim, H.Y.; Cheong, J. Endoplasmic reticulum stress induced by hepatitis B virus X protein enhances cyclo-oxygenase 2 expression via activating transcription factor 4. Biochem. J. 2011, 435, 431–439. [Google Scholar] [CrossRef]
- Lukas, J.; Pospech, J.; Oppermann, C.; Hund, C.; Iwanov, K.; Pantoom, S.; Petters, J.; Frech, M.; Seemann, S.; Thiel, F.G.; et al. Role of endoplasmic reticulum stress and protein misfolding in disorders of the liver and pancreas. Adv. Med. Sci. 2019, 64, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Flessa, C.M.; Kyrou, I.; Nasiri-Ansari, N.; Kaltsas, G.; Papavassiliou, A.G.; Kassi, E.; Randeva, H.S. Endoplasmic Reticulum Stress and Autophagy in the Pathogenesis of Non-alcoholic Fatty Liver Disease (NAFLD): Current Evidence and Perspectives. Curr. Obes. Rep. 2021, 10, 134–161. [Google Scholar] [CrossRef]
- Waris, G.; Tardif, K.D.; Siddiqui, A. Endoplasmic reticulum (ER) stress: Hepatitis C virus induces an ER-nucleus signal transduction pathway and activates NF-kappaB and STAT-3. Biochem. Pharmacol. 2002, 64, 1425–1430. [Google Scholar] [CrossRef]
- Villalobos-Labra, R.; Subiabre, M.; Toledo, F.; Pardo, F.; Sobrevia, L. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol. Asp. Med. 2019, 66, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Luan, J.; Lv, X. Inhibition of Endoplasmic Reticulum Stress Attenuated Ethanol-Induced Exosomal miR-122 and Acute Liver Injury in Mice. Alcohol Alcohol. 2019, 54, 465–471. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, X.; Wu, J.L.; Fu, L.; Liu, K.; Liu, D.; Chen, G.G.; Lai, P.B.; Wong, N.; Yu, J. O-GlcNAc transferase promotes fatty liver-associated liver cancer through inducing palmitic acid and activating endoplasmic reticulum stress. J. Hepatol. 2017, 67, 310–320. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Li, S.; Huang, M.; Xiong, Y.; Zhang, Q.; Ye, L.; Liu, J.; Zhu, X.; Sun, R.; Guo, Y. The roles of endoplasmic reticulum overload response induced by HCV and NS4B protein in human hepatocyte viability and virus replication. PLoS ONE 2015, 10, e0123190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev.. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef]
- Roy, B.; Lee, A.S. The mammalian endoplasmic reticulum stress response element consists of an evolutionarily conserved tripartite structure and interacts with a novel stress-inducible complex. Nucleic Acids Res. 1999, 27, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef] [Green Version]
- Kokame, K.; Kato, H.; Miyata, T. Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J. Biol. Chem. 2001, 276, 9199–9205. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Yoshida, H.; Kokame, K.; Kaufman, R.J.; Mori, K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 2004, 136, 343–350. [Google Scholar] [CrossRef]
- Oyadomari, S.; Harding, H.P.; Zhang, Y.; Oyadomari, M.; Ron, D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008, 7, 520–532. [Google Scholar] [CrossRef] [Green Version]
- Morita, S.; Takeshima, K.; Ariyasu, H.; Furukawa, Y.; Kishimoto, S.; Tsuji, T.; Uraki, S.; Mishima, H.; Kinoshita, A.; Takahashi, Y.; et al. Expression of unfolded protein response markers in the pheochromocytoma with Waardenburg syndrome: A case report. BMC Endocr. Disord. 2020, 20, 90. [Google Scholar] [CrossRef] [PubMed]
- Stroberg, W.; Eilertsen, J.; Schnell, S. Information processing by endoplasmic reticulum stress sensors. J. R. Soc. Interface 2019, 16, 20190288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Pan, E.; Zhu, J.; Xu, L.; Chen, X.; Li, J.; Liang, L.; Hu, Y.; Xia, J.; Chen, J.; et al. Cisplatin Enhances Hepatitis B Virus Replication and PGC-1α Expression through Endoplasmic Reticulum Stress. Sci. Rep. 2018, 8, 3496. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, S.; Aydin, Y.; Wu, T. Integrated stress response in hepatitis C promotes Nrf2-related chaperone-mediated autophagy: A novel mechanism for host-microbe survival and HCC development in liver cirrhosis. Semin. Cell Dev. Biol. 2020, 101, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Suwanmanee, Y.; Wada, M.; Ueda, K. Functional roles of GRP78 in hepatitis B virus infectivity and antigen secretion. Microbiol. Immunol. 2021, 65, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef] [PubMed]
- Korennykh, A.; Walter, P. Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2012, 28, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Kang, T.I.; So, J.S. Roles of XBP1s in Transcriptional Regulation of Target Genes. Biomedicines 2021, 9, 791. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, L.; Li, F.; Li, S.; Zhang, M.; Wang, F.; Lv, X.; Liu, Q. Effect of Different Exercise Intensities on Hepatocyte Apoptosis in HFD-Induced NAFLD in Rats: The Possible Role of Endoplasmic Reticulum Stress through the Regulation of the IRE1/JNK and eIF2α/CHOP Signal Pathways. Oxidative Med. Cell. Longev. 2021, 2021, 6378568. [Google Scholar] [CrossRef]
- Liu, C.M.; Zheng, G.H.; Ming, Q.L.; Sun, J.M.; Cheng, C. Protective effect of quercetin on lead-induced oxidative stress and endoplasmic reticulum stress in rat liver via the IRE1/JNK and PI3K/Akt pathway. Free Radic. Res. 2013, 47, 192–201. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [Green Version]
- Yi, S.; Chen, K.; Zhang, L.; Shi, W.; Zhang, Y.; Niu, S.; Jia, M.; Cong, B.; Li, Y. Endoplasmic Reticulum Stress Is Involved in Stress-Induced Hypothalamic Neuronal Injury in Rats via the PERK-ATF4-CHOP and IRE1-ASK1-JNK Pathways. Front. Cell. Neurosci. 2019, 13, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, H.; Zhu, C.; Wei, W.; Lv, X.; Yu, Q.; Deng, X.; Ci, X. Enhanced Keap1-Nrf2/Trx-1 axis by daphnetin protects against oxidative stress-driven hepatotoxicity via inhibiting ASK1/JNK and Txnip/NLRP3 inflammasome activation. Phytomed. Int. J. Phytother. Phytopharm. 2020, 71, 153241. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, D.; Li, Y.; Qiao, H.; Shan, Z. Ketamine enhances autophagy and endoplasmic reticulum stress in rats and SV-HUC-1 cells via activating IRE1-TRAF2-ASK1-JNK pathway. Cell Cycle 2021, 20, 1907–1922. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Zhang, Y.; Ji, W.; Wang, L.; Lee, S.C. Periplogenin Activates ROS-ER Stress Pathway to Trigger Apoptosis via BIP-eIF2α- CHOP and IRE1α-ASK1-JNK Signaling Routes. Anti-Cancer Agents Med. Chem. 2021, 21, 61–70. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, S.Y.; Kim, M.; Cheon, C.; Ko, S.G. Kaempferol induces autophagic cell death via IRE1-JNK-CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis. 2018, 9, 875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Feng, Y.; Huang, H.; Cui, L.; Li, F. PM2.5 exposure induces reproductive injury through IRE1/JNK/autophagy signaling in male rats. Ecotoxicol. Environ. Saf. 2021, 211, 111924. [Google Scholar] [CrossRef]
- Quan, J.H.; Gao, F.F.; Lee, M.; Yuk, J.M.; Cha, G.H.; Chu, J.Q.; Wang, H.; Lee, Y.H. Involvement of endoplasmic reticulum stress response and IRE1-mediated ASK1/JNK/Mcl-1 pathways in silver nanoparticle-induced apoptosis of human retinal pigment epithelial cells. Toxicology 2020, 442, 152540. [Google Scholar] [CrossRef]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell. Mol. Life Sci. CMLS 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Xiao-Wei, L.; Juan, W.; Xiu-Juan, L.; Nian-Yun, Z.; Lei, S. HIIT and MICT attenuate high-fat diet-induced hepatic lipid accumulation and ER stress via the PERK-ATF4-CHOP signaling pathway. J. Physiol. Biochem. 2022, 78, 641–652. [Google Scholar] [CrossRef]
- Cao, J.; Dai, D.L.; Yao, L.; Yu, H.H.; Ning, B.; Zhang, Q.; Chen, J.; Cheng, W.H.; Shen, W.; Yang, Z.X. Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol. Cell. Biochem. 2012, 364, 115–129. [Google Scholar] [CrossRef]
- Guo, Y.; Guo, R.; Su, Y.; Fu, J.; Wang, S.; Kong, Y.; Wu, C.; Wang, J.; Tan, C.; Mo, C.; et al. The PERK/eIF2α/ATF4/CHOP pathway plays a role in regulating monocrotaline-induced endoplasmic reticulum stress in rat liver. Res. Vet. Sci. 2020, 130, 237–239. [Google Scholar] [CrossRef]
- Jo, H.J.; Yang, J.W.; Park, J.H.; Choi, E.S.; Lim, C.S.; Lee, S.; Han, C.Y. Endoplasmic Reticulum Stress Increases DUSP5 Expression via PERK-CHOP Pathway, Leading to Hepatocyte Death. Int. J. Mol. Sci. 2019, 20, 4369. [Google Scholar] [CrossRef]
- Ben Mosbah, I.; Duval, H.; Mbatchi, S.F.; Ribault, C.; Grandadam, S.; Pajaud, J.; Morel, F.; Boudjema, K.; Compagnon, P.; Corlu, A. Intermittent selective clamping improves rat liver regeneration by attenuating oxidative and endoplasmic reticulum stress. Cell Death Dis. 2014, 5, e1107. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.B.; Darko, C.; Alonso, L.C. Intersection of the ATF6 and XBP1 ER stress pathways in mouse islet cells. J. Biol. Chem. 2020, 295, 14164–14177. [Google Scholar] [CrossRef]
- Pahl, H.L.; Baeuerle, P.A. The ER-overload response: Activation of NF-kappa B. Trends Biochem. Sci. 1997, 22, 63–67. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Kouri, G.; Wollheim, C.B. ER stress and SREBP-1 activation are implicated in beta-cell glucolipotoxicity. J. Cell Sci. 2005, 118, 3905–3915. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, A. Virus-induced ER stress and the unfolded protein response. Front. Plant Sci. 2012, 3, 293. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; HuangFu, W.C.; Kumar, K.G.; Qian, J.; Casey, J.P.; Hamanaka, R.B.; Grigoriadou, C.; Aldabe, R.; Diehl, J.A.; Fuchs, S.Y. Virus-induced unfolded protein response attenuates antiviral defenses via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe 2009, 5, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wei, Z.; Cheng, B.; Li, J.; He, Y.; Lan, T.; Kemper, T.; Lin, Y.; Jiang, B.; Jiang, Y.; et al. Endoplasmic reticulum stress promotes HBV production by enhancing use of the autophagosome/multivesicular body axis. Hepatology 2022, 75, 438–454. [Google Scholar] [CrossRef]
- Tardif, K.D.; Mori, K.; Kaufman, R.J.; Siddiqui, A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 2004, 279, 17158–17164. [Google Scholar] [CrossRef]
- Isler, J.A.; Skalet, A.H.; Alwine, J.C. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 2005, 79, 6890–6899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Gao, B.; Ye, L.; Han, X.; Wang, W.; Kong, L.; Fang, X.; Zeng, Y.; Zheng, H.; Li, S.; et al. Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of unfolded protein response. Virus Res. 2007, 124, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Pavio, N.; Romano, P.R.; Graczyk, T.M.; Feinstone, S.M.; Taylor, D.R. Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2alpha kinase PERK. J. Virol. 2003, 77, 3578–3585. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Feng, Z.; He, B. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma(1)34.5 protein. J. Virol. 2005, 79, 1379–1388. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xia, Y.; Cheng, X.; Kleiner, D.E.; Hewitt, S.M.; Sproch, J.; Li, T.; Zhuang, H.; Liang, T.J. Hepatitis B Surface Antigen Activates Unfolded Protein Response in Forming Ground Glass Hepatocytes of Chronic Hepatitis B. Viruses 2019, 11, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos-Ocampo, W.A.; Navas, M.C.; Buist-Homan, M.; Faber, K.N.; Daemen, T.; Moshage, H. Hepatitis C Virus Proteins Core and NS5A Are Highly Sensitive to Oxidative Stress-Induced Degradation after eIF2α/ATF4 Pathway Activation. Viruses 2020, 12, 425. [Google Scholar] [CrossRef] [Green Version]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Fung, T.S.; Huang, M.; Fang, S.G.; Zhong, Y.; Liu, D.X. Upregulation of CHOP/GADD153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J. Virol. 2013, 87, 8124–8134. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jiang, W.; Niu, Q.; Sun, Y.; Meng, C.; Tan, L.; Song, C.; Qiu, X.; Liao, Y.; Ding, C. eIF2α-CHOP-BCl-2/JNK and IRE1α-XBP1/JNK signaling promote apoptosis and inflammation and support the proliferation of Newcastle disease virus. Cell Death Dis. 2019, 10, 891. [Google Scholar] [CrossRef]
- Isobe, T.; Tange, S.; Tasaki, H.; Kanamori, K.; Kato, A.; Nakanishi, A. Upregulation of CHOP participates in caspase activation and virus release in human astrovirus-infected cells. J. Gen. Virol. 2019, 100, 778–792. [Google Scholar] [CrossRef]
- Zhang, F.; Moon, A.; Childs, K.; Goodbourn, S.; Dixon, L.K. The African swine fever virus DP71L protein recruits the protein phosphatase 1 catalytic subunit to dephosphorylate eIF2alpha and inhibits CHOP induction but is dispensable for these activities during virus infection. J. Virol. 2010, 84, 10681–10689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, L.; Thomas, S.; Herchenröder, O.; Pützer, B.M.; Schaefer, S. Hepatitis E virus ORF2 protein activates the pro-apoptotic gene CHOP and anti-apoptotic heat shock proteins. PLoS ONE 2011, 6, e25378. [Google Scholar] [CrossRef]
- Joyce, M.A.; Walters, K.A.; Lamb, S.E.; Yeh, M.M.; Zhu, L.F.; Kneteman, N.; Doyle, J.S.; Katze, M.G.; Tyrrell, D.L. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 2009, 5, e1000291. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Liu, Y.; Liu, X.; Li, T.; Xin, Z.; Zhu, X.; Wu, X.; Liu, Y. HBV suppresses thapsigargin-induced apoptosis via inhibiting CHOP expression in hepatocellular carcinoma cells. Oncol. Lett. 2017, 14, 4403–4409. [Google Scholar] [CrossRef]
- Churin, Y.; Roderfeld, M.; Stiefel, J.; Würger, T.; Schröder, D.; Matono, T.; Mollenkopf, H.J.; Montalbano, R.; Pompaiah, M.; Reifenberg, K.; et al. Pathological impact of hepatitis B virus surface proteins on the liver is associated with the host genetic background. PLoS ONE 2014, 9, e90608. [Google Scholar] [CrossRef] [Green Version]
- Tardif, K.D.; Mori, K.; Siddiqui, A. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J. Virol. 2002, 76, 7453–7459. [Google Scholar] [CrossRef] [Green Version]
- Pasqual, G.; Burri, D.J.; Pasquato, A.; de la Torre, J.C.; Kunz, S. Role of the host cell’s unfolded protein response in arenavirus infection. J. Virol. 2011, 85, 1662–1670. [Google Scholar] [CrossRef] [Green Version]
- Galindo, I.; Hernáez, B.; Muñoz-Moreno, R.; Cuesta-Geijo, M.A.; Dalmau-Mena, I.; Alonso, C. The ATF6 branch of unfolded protein response and apoptosis are activated to promote African swine fever virus infection. Cell Death Dis. 2012, 3, e341. [Google Scholar] [CrossRef] [Green Version]
- Trujillo-Alonso, V.; Maruri-Avidal, L.; Arias, C.F.; López, S. Rotavirus infection induces the unfolded protein response of the cell and controls it through the nonstructural protein NSP3. J. Virol. 2011, 85, 12594–12604. [Google Scholar] [CrossRef]
- Tan, Z.; Zhang, W.; Sun, J.; Fu, Z.; Ke, X.; Zheng, C.; Zhang, Y.; Li, P.; Liu, Y.; Hu, Q.; et al. ZIKV infection activates the IRE1-XBP1 and ATF6 pathways of unfolded protein response in neural cells. J. Neuroinflamm. 2018, 15, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.W.; Wang, S.S.; Hung, C.H.; Hung, Y.H.; Lin, C.L.; Chang, P.J. The Epstein-Barr Virus Lytic Protein BMLF1 Induces Upregulation of GRP78 Expression through ATF6 Activation. Int. J. Mol. Sci. 2021, 22, 4024. [Google Scholar] [CrossRef]
- Tardif, K.D.; Siddiqui, A. Cell surface expression of major histocompatibility complex class I molecules is reduced in hepatitis C virus subgenomic replicon-expressing cells. J. Virol. 2003, 77, 11644–11650. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Yang, J.; Hu, Q.; Mao, J.; Yang, Q.; Chen, H.; Li, D.; Li, P.; Duan, L.; Wang, B.; et al. SIP1 serves a role in HBx-induced liver cancer growth and metastasis. Int. J. Oncol. 2019, 55, 1019–1032. [Google Scholar] [CrossRef] [Green Version]
- Su, I.J.; Wang, H.C.; Wu, H.C.; Huang, W.Y. Ground glass hepatocytes contain pre-S mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J. Gastroenterol. Hepatol. 2008, 23, 1169–1174. [Google Scholar] [CrossRef]
- Baudi, I.; Isogawa, M.; Moalli, F.; Onishi, M.; Kawashima, K.; Ishida, Y.; Tateno, C.; Sato, Y.; Harashima, H.; Ito, H.; et al. Interferon signaling suppresses the unfolded protein response and induces cell death in hepatocytes accumulating hepatitis B surface antigen. PLoS Pathog. 2021, 17, e1009228. [Google Scholar] [CrossRef]
- Cho, H.K.; Kim, S.Y.; Seong, J.K.; Cheong, J. Hepatitis B virus X increases immune cell recruitment by induction of chemokine SDF-1. FEBS Lett. 2014, 588, 733–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, K.; Sudo, T.; Morishima, N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J. Cell Biol. 2005, 169, 555–560. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.M.; Lee, S.Y.; Kim, B.J. Naturally Occurring Hepatitis B Virus Mutations Leading to Endoplasmic Reticulum Stress and Their Contribution to the Progression of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 597. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Kim, H.; Lee, S.A.; Won, Y.S.; Kim, H.I.; Inn, K.S.; Kim, B.J. Upregulation of endoplasmic reticulum stress and reactive oxygen species by naturally occurring mutations in hepatitis B virus core antigen. J. Gen. Virol. 2015, 96, 1850–1854. [Google Scholar] [CrossRef]
- Lin, W.L.; Hung, J.H.; Huang, W. Association of the Hepatitis B Virus Large Surface Protein with Viral Infectivity and Endoplasmic Reticulum Stress-mediated Liver Carcinogenesis. Cells 2020, 9, 2052. [Google Scholar] [CrossRef] [PubMed]
- Paridaens, A.; Laukens, D.; Vandewynckel, Y.P.; Coulon, S.; Van Vlierberghe, H.; Geerts, A.; Colle, I. Endoplasmic reticulum stress and angiogenesis: Is there an interaction between them? Liver Int. Off. J. Int. Assoc. Study Liver 2014, 34, e10–e18. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Cacciola, I.; Saffioti, F.; Raimondo, G. Hepatitis B virus PreS/S gene variants: Pathobiology and clinical implications. J. Hepatol. 2014, 61, 408–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.T.; Jeng, L.B.; Chan, W.L.; Su, I.J.; Teng, C.F. Hepatitis B Virus Pre-S Gene Deletions and Pre-S Deleted Proteins: Clinical and Molecular Implications in Hepatocellular Carcinoma. Viruses 2021, 13, 862. [Google Scholar] [CrossRef]
- Montalbano, R.; Honrath, B.; Wissniowski, T.T.; Elxnat, M.; Roth, S.; Ocker, M.; Quint, K.; Churin, Y.; Roederfeld, M.; Schroeder, D.; et al. Exogenous hepatitis B virus envelope proteins induce endoplasmic reticulum stress: Involvement of cannabinoid axis in liver cancer cells. Oncotarget 2016, 7, 20312–20323. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.C.; Wu, H.C.; Chen, C.F.; Fausto, N.; Lei, H.Y.; Su, I.J. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am. J. Pathol. 2003, 163, 2441–2449. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Jensen, G.; Yen, T.S. Activation of hepatitis B virus S promoter by the viral large surface protein via induction of stress in the endoplasmic reticulum. J. Virol. 1997, 71, 7387–7392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, C.T.; Tillmann, H.L.; Manns, M.P.; Trautwein, C. The pre-S region determines the intracellular localization and appearance of hepatitis B virus. Hepatology 1999, 30, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, L.; Lu, Y.; Wei, K.P.; Liu, Z.X.; Xiao, Y.W.; Ding, F.; Zhuang, H.; Li, J. Correlation between serum HBV DNA level and HBsAg titer in HBeAg-positive pregnant women and impact of genomic variability of hepatitis B virus pre S/S regions on their correlations. Zhonghua Gan Zang Bing Za Zhi = Zhonghua Ganzangbing Zazhi = Chin. J. Hepatol. 2018, 26, 579–584. [Google Scholar] [CrossRef]
- Wang, H.C.; Huang, W.; Lai, M.D.; Su, I.J. Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci. 2006, 97, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Ren, Y.L.; Fu, H.J.; Zou, L.; Yang, Y.; Chen, Z. Hepatitis B Virus Middle Protein Enhances IL-6 Production via p38 MAPK/NF-κB Pathways in an ER Stress-Dependent Manner. PLoS ONE 2016, 11, e0159089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollicino, T.; Amaddeo, G.; Restuccia, A.; Raffa, G.; Alibrandi, A.; Cutroneo, G.; Favaloro, A.; Maimone, S.; Squadrito, G.; Raimondo, G. Impact of hepatitis B virus (HBV) preS/S genomic variability on HBV surface antigen and HBV DNA serum levels. Hepatology 2012, 56, 434–443. [Google Scholar] [CrossRef]
- Hildt, E.; Munz, B.; Saher, G.; Reifenberg, K.; Hofschneider, P.H. The PreS2 activator MHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling in transgenic mice. EMBO J. 2002, 21, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Maiers, J.L.; Rui, Y.; Jiang, X.; Guleng, B.; Ren, J. Apolipoprotein H drives hepatitis B surface antigen retention and endoplasmic reticulum stress during hepatitis B virus infection. Int. J. Biochem. Cell Biol. 2021, 131, 105906. [Google Scholar] [CrossRef]
- Liang, Y.J.; Teng, W.; Chen, C.L.; Sun, C.P.; Teng, R.D.; Huang, Y.H.; Liang, K.H.; Chen, Y.W.; Lin, C.C.; Su, C.W.; et al. Clinical Implications of HBV PreS/S Mutations and the Effects of PreS2 Deletion on Mitochondria, Liver Fibrosis, and Cancer Development. Hepatology 2021, 74, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.F. Hepatitis B virus pre-S/S variants in liver diseases. World J. Gastroenterol. 2018, 24, 1507–1520. [Google Scholar] [CrossRef]
- Li, Y.W.; Yang, F.C.; Lu, H.Q.; Zhang, J.S. Hepatocellular carcinoma and hepatitis B surface protein. World J. Gastroenterol. 2016, 22, 1943–1952. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Bühler, S.; Bartenschlager, R. Chronic viral hepatitis and its association with liver cancer. Biol. Chem. 2017, 398, 817–837. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.X.; Ye, S.S.; Hong, Y.X.; Chen, Y.; Wang, B.; Lin, X.J.; Lin, X. Hepatitis B Virus Small Envelope Protein Promotes Hepatocellular Carcinoma Angiogenesis via Endoplasmic Reticulum Stress Signaling To Upregulate the Expression of Vascular Endothelial Growth Factor A. J. Virol. 2022, 96, e0197521. [Google Scholar] [CrossRef]
- Liepelt, A.; Tacke, F. Stromal cell-derived factor-1 (SDF-1) as a target in liver diseases. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G203–G209. [Google Scholar] [CrossRef]
- Li, J.; He, J.; Fu, Y.; Hu, X.; Sun, L.Q.; Huang, Y.; Fan, X. Hepatitis B virus X protein inhibits apoptosis by modulating endoplasmic reticulum stress response. Oncotarget 2017, 8, 96027–96034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muroyama, R.; Nakagawa, R.; Matsubara, Y.; Hirata, Y.; Omata, M.; Shirasawa, H.; Kato, N. Fusion HBx from HBV integrant affects hepatocarcinogenesis through deregulation of ER stress response. Virus Res. 2022, 315, 198787. [Google Scholar] [CrossRef]
- He, C.; Qiu, Y.; Han, P.; Chen, Y.; Zhang, L.; Yuan, Q.; Zhang, T.; Cheng, T.; Yuan, L.; Huang, C.; et al. ER stress regulating protein phosphatase 2A-B56γ, targeted by hepatitis B virus X protein, induces cell cycle arrest and apoptosis of hepatocytes. Cell Death Dis. 2018, 9, 762. [Google Scholar] [CrossRef] [Green Version]
- Ahodantin, J.; Lekbaby, B.; Bou Nader, M.; Soussan, P.; Kremsdorf, D. Hepatitis B virus X protein enhances the development of liver fibrosis and the expression of genes associated with epithelial-mesenchymal transitions and tumor progenitor cells. Carcinogenesis 2020, 41, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.H.; Su, I.J.; Lei, H.Y.; Wang, H.C.; Lin, W.C.; Chang, W.T.; Huang, W.; Chang, W.C.; Chang, Y.S.; Chen, C.C.; et al. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J. Biol. Chem. 2004, 279, 46384–46392. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.S.; Chan, H.L.; Leung, W.K.; To, K.F.; Go, M.Y.; Chan, J.Y.; Liew, C.T.; Sung, J.J. Expression of HBx and COX-2 in chronic hepatitis B, cirrhosis and hepatocellular carcinoma: Implication of HBx in upregulation of COX-2. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2004, 17, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, H.; Osaki, Y. Liver Cirrhosis: Evaluation, Nutritional Status, and Prognosis. Mediat. Inflamm. 2015, 2015, 872152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunduz, F.; Aboulnasr, F.M.; Chandra, P.K.; Hazari, S.; Poat, B.; Baker, D.P.; Balart, L.A.; Dash, S. Free fatty acids induce ER stress and block antiviral activity of interferon alpha against hepatitis C virus in cell culture. Virol. J. 2012, 9, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhaveri, R.; Qiang, G.; Diehl, A.M. Domain 3 of hepatitis C virus core protein is sufficient for intracellular lipid accumulation. J. Infect. Dis. 2009, 200, 1781–1788. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Mu, M.; Chen, H.; Zhang, G.; Yang, Y.; Chu, J.; Li, Y.; Yang, F.; Lin, S. Hepatocyte steatosis inhibits hepatitis B virus secretion via induction of endoplasmic reticulum stress. Mol. Cell. Biochem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Shin, H.J.; Kim, K.; Choi, H.M.; Rhee, S.H.; Moon, H.B.; Kim, H.H.; Yang, U.S.; Yu, D.Y.; Cheong, J. Hepatitis B virus X protein induces hepatic steatosis via transcriptional activation of SREBP1 and PPARgamma. Gastroenterology 2007, 132, 1955–1967. [Google Scholar] [CrossRef]
- Ding, L.Y.; Liang, L.Z.; Zhao, Y.X.; Yang, Y.N.; Liu, F.; Ding, Q.R.; Luo, L.J. Porphyromonas gingivalis-derived lipopolysaccharide causes excessive hepatic lipid accumulation via activating NF-κB and JNK signaling pathways. Oral Dis. 2019, 25, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, M.; Mathews, S.; Makarov, E.; Petrosyan, A.; Kharbanda, K.K.; Kidambi, S.; Poluektova, L.Y.; Casey, C.A.; Osna, N.A. Acetaldehyde suppresses HBV-MHC class I complex presentation on hepatocytes via induction of ER stress and Golgi fragmentation. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G432–G442. [Google Scholar] [CrossRef] [PubMed]
- Gawlik, K.; Gallay, P.A. HCV core protein and virus assembly: What we know without structures. Immunol. Res. 2014, 60, 1–10. [Google Scholar] [CrossRef]
- Asselah, T.; Bièche, I.; Mansouri, A.; Laurendeau, I.; Cazals-Hatem, D.; Feldmann, G.; Bedossa, P.; Paradis, V.; Martinot-Peignoux, M.; Lebrec, D.; et al. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J. Pathol. 2010, 221, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Sato, N.; Kikuchi, J.; Kakinuma, H.; Okawa, J.; Masuyama, Y.; Iwasa, S.; Irokawa, H.; Hwang, G.W.; Naganuma, A.; et al. Immature Core protein of hepatitis C virus induces an unfolded protein response through inhibition of ERAD-L in a yeast model system. Genes Cells Devoted Mol. Cell. Mech. 2017, 22, 160–173. [Google Scholar] [CrossRef] [Green Version]
- Benali-Furet, N.L.; Chami, M.; Houel, L.; De Giorgi, F.; Vernejoul, F.; Lagorce, D.; Buscail, L.; Bartenschlager, R.; Ichas, F.; Rizzuto, R.; et al. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 2005, 24, 4921–4933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von dem Bussche, A.; Machida, R.; Li, K.; Loevinsohn, G.; Khander, A.; Wang, J.; Wakita, T.; Wands, J.R.; Li, J. Hepatitis C virus NS2 protein triggers endoplasmic reticulum stress and suppresses its own viral replication. J. Hepatol. 2010, 53, 797–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos-Ocampo, W.A.; Daemen, T.; Buist-Homan, M.; Faber, K.N.; Navas, M.C.; Moshage, H. Hepatitis C virus core or NS3/4A protein expression preconditions hepatocytes against oxidative stress and endoplasmic reticulum stress. Redox Rep. Commun. Free Radic. Res. 2019, 24, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Gao, B.; Ye, L.; Kong, L.; Jing, W.; Yang, X.; Wu, Z.; Ye, L. Hepatitis C virus non-structural protein NS4B can modulate an unfolded protein response. J. Microbiol. 2005, 43, 529–536. [Google Scholar] [PubMed]
- Meng, Z.; Liu, Q.; Sun, F.; Qiao, L. Hepatitis C virus nonstructural protein 5A perturbs lipid metabolism by modulating AMPK/SREBP-1c signaling. Lipids Health Dis. 2019, 18, 191. [Google Scholar] [CrossRef] [Green Version]
- Mishima, K.; Sakamoto, N.; Sekine-Osajima, Y.; Nakagawa, M.; Itsui, Y.; Azuma, S.; Kakinuma, S.; Kiyohashi, K.; Kitazume, A.; Tsuchiya, K.; et al. Cell culture and in vivo analyses of cytopathic hepatitis C virus mutants. Virology 2010, 405, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.L.; Wan, Y.; Shen, W.; Cao, J.; Sun, Z.X.; Yu, H.H.; Zhang, Q.; Cheng, W.H.; Chen, J.; Ning, B. Endoplasmic reticulum stress leads to lipid accumulation through upregulation of SREBP-1c in normal hepatic and hepatoma cells. Mol. Cell. Biochem. 2013, 381, 127–137. [Google Scholar] [CrossRef]
- Selby, M.; Erickson, A.; Dong, C.; Cooper, S.; Parham, P.; Houghton, M.; Walker, C.M. Hepatitis C virus envelope glycoprotein E1 originates in the endoplasmic reticulum and requires cytoplasmic processing for presentation by class I MHC molecules. J. Immunol. 1999, 162, 669–676. [Google Scholar] [PubMed]
- Ciccaglione, A.R.; Marcantonio, C.; Tritarelli, E.; Equestre, M.; Vendittelli, F.; Costantino, A.; Geraci, A.; Rapicetta, M. Activation of the ER stress gene gadd153 by hepatitis C virus sensitizes cells to oxidant injury. Virus Res. 2007, 126, 128–138. [Google Scholar] [CrossRef]
- Nierenberg, A.A.; Ghaznavi, S.A.; Sande Mathias, I.; Ellard, K.K.; Janos, J.A.; Sylvia, L.G. Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1 Alpha as a Novel Target for Bipolar Disorder and Other Neuropsychiatric Disorders. Biol. Psychiatry 2018, 83, 761–769. [Google Scholar] [CrossRef]
- Hesler, S.; Angeliadis, M.; Husain, B.; Cole, J.L. Contribution of dsRBD2 to PKR Activation. ACS Omega 2021, 6, 11367–11374. [Google Scholar] [CrossRef] [PubMed]
- He, B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006, 13, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, H.; Zhang, J.; Sha, Y.; Wu, F.; Wen, S.; He, L.; Sheng, L.; You, Q.; Shi, M.; et al. SPHK1 deficiency protects mice from acetaminophen-induced ER stress and mitochondrial permeability transition. Cell Death Differ. 2020, 27, 1924–1937. [Google Scholar] [CrossRef]
- Aizawa, S.; Okamoto, T.; Sugiyama, Y.; Kouwaki, T.; Ito, A.; Suzuki, T.; Ono, C.; Fukuhara, T.; Yamamoto, M.; Okochi, M.; et al. TRC8-dependent degradation of hepatitis C virus immature core protein regulates viral propagation and pathogenesis. Nat. Commun. 2016, 7, 11379. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, N.S.; McIntyre, A.B.R.; Mattocks, M.D.; Holley, C.L.; Lazear, H.M.; Mason, C.E.; Horner, S.M. Altered m(6)A Modification of Specific Cellular Transcripts Affects Flaviviridae Infection. Mol. Cell 2020, 77, 542–555.e548. [Google Scholar] [CrossRef]
- Bassendine, M.F.; Sheridan, D.A.; Bridge, S.H.; Felmlee, D.J.; Neely, R.D. Lipids and HCV. Semin. Immunopathol. 2013, 35, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Takaki, H.; Akazawa, Y.; Kido, Y.; Morishita, M.; Honda, T.; Shibata, H.; Miuma, S.; Miyaaki, H.; Taura, N.; Kondo, H.; et al. Hepatitis C Virus Infection Increases c-Jun N-Terminal Kinase (JNK) Phosphorylation and Accentuates Hepatocyte Lipoapoptosis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 4526–4532. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Mao, Y.; Liu, M.; Luo, R.; Jiang, R.; Guo, F. The active nuclear form of SREBP1 amplifies ER stress and autophagy via regulation of PERK. FEBS J. 2020, 287, 2348–2366. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Kaplowitz, N.; Lebeaupin, C.; Kroemer, G.; Kaufman, R.J.; Malhi, H.; Ren, J. Endoplasmic reticulum stress in liver diseases. Hepatology 2022. [Google Scholar] [CrossRef] [PubMed]
- Ji, C. Dissection of endoplasmic reticulum stress signaling in alcoholic and non-alcoholic liver injury. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. 1), S16–S24. [Google Scholar] [CrossRef] [Green Version]
- Rutkowski, D.T. Liver function and dysfunction—A unique window into the physiological reach of ER stress and the unfolded protein response. FEBS J. 2019, 286, 356–378. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Qiao, S.; Xiang, Y.; Cui, M.; Yao, X.; Lin, R.; Zhang, X. Endoplasmic reticulum stress: Multiple regulatory roles in hepatocellular carcinoma. Biomed. Pharmacother. 2021, 142, 112005. [Google Scholar] [CrossRef]
- Ke, P.Y.; Chen, S.S. Hepatitis C virus and cellular stress response: Implications to molecular pathogenesis of liver diseases. Viruses 2012, 4, 2251–2290. [Google Scholar] [CrossRef]
- Chan, S.W. Unfolded protein response in hepatitis C virus infection. Front. Microbiol. 2014, 5, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawal, R.K.; Konreddy, A.K.; Chu, C.K. Mechanism of Adefovir, Tenofovir and Entecavir Resistance: Molecular Modeling Studies of How A Novel Anti-HBV Agent (FMCA) Can Overcome the Drug Resistance. Curr. Med. Chem. 2015, 22, 3922–3932. [Google Scholar] [CrossRef]
- Chen, H.; Mu, M.; Liu, Q.; Hu, H.; Tian, C.; Zhang, G.; Li, Y.; Yang, F.; Lin, S. Hepatocyte Endoplasmic Reticulum Stress Inhibits Hepatitis B Virus Secretion and Delays Intracellular Hepatitis B Virus Clearance After Entecavir Treatment. Front. Med. 2020, 7, 589040. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, L.; Li, G.; Libin, P.; Piampongsant, S.; Vandamme, A.M.; Theys, K. Genetic Diversity and Selective Pressure in Hepatitis C Virus Genotypes 1-6: Significance for Direct-Acting Antiviral Treatment and Drug Resistance. Viruses 2015, 7, 5018–5039. [Google Scholar] [CrossRef] [Green Version]
- Lemon, S.M. Induction and evasion of innate antiviral responses by hepatitis C virus. J. Biol. Chem. 2010, 285, 22741–22747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nistal-Villán, E.; Argemi, J.; de Jaime-Soguero, A.; Ferrero, R.; di Scala, M.; Rodriguez-Garcia, E.; Coll, A.; Rius-Rocabert, S.; Prieto, J.; González-Aseguinolaza, G.; et al. Linking the Expression of Therapeutic Genes to Unfolded Protein Response: A New Option for Anti-Hepatitis B Virus Gene Therapy. Hum. Gene Ther. 2021, 32, 341–348. [Google Scholar] [CrossRef]
- Yao, W.; Cai, H.; Li, X.; Li, T.; Hu, L.; Peng, T. Endoplasmic reticulum stress links hepatitis C virus RNA replication to wild-type PGC-1α/liver-specific PGC-1α upregulation. J. Virol. 2014, 88, 8361–8374. [Google Scholar] [CrossRef] [Green Version]
- Wang, K. Autophagy and apoptosis in liver injury. Cell Cycle 2015, 14, 1631–1642. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Qi, B.; Gu, Y.; Xu, F.; Du, H.; Li, X.; Fang, W. Porcine Circovirus 2 Deploys PERK Pathway and GRP78 for Its Enhanced Replication in PK-15 Cells. Viruses 2016, 8, 56. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, Y.; Imajo, K.; Yoneda, M.; Tomeno, W.; Ogawa, Y.; Kirikoshi, H.; Funakoshi, K.; Ikeda, M.; Kato, N.; Nakajima, A.; et al. Unfolded protein response pathways regulate Hepatitis C virus replication via modulation of autophagy. Biochem. Biophys. Res. Commun. 2013, 432, 326–332. [Google Scholar] [CrossRef]


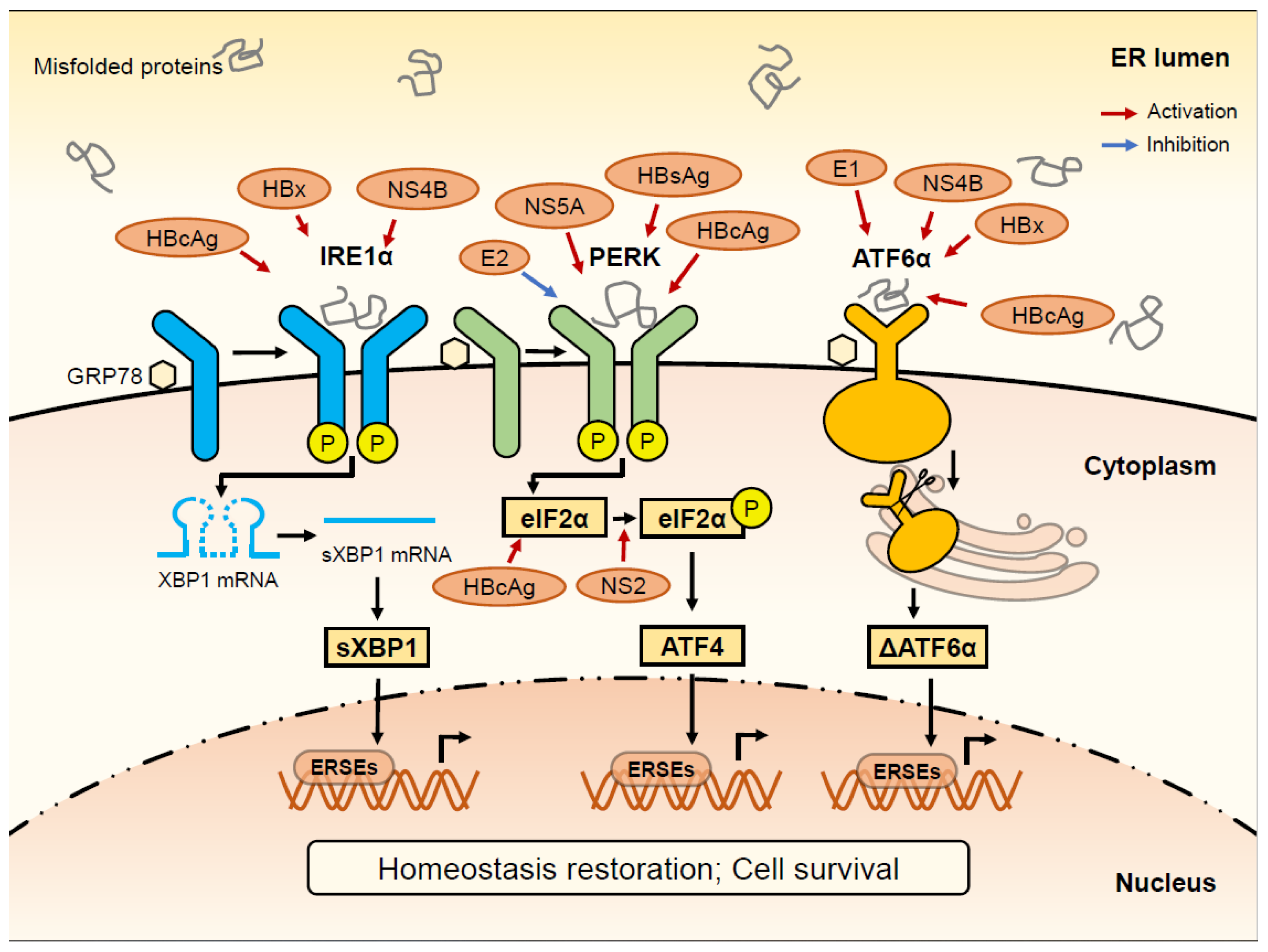{kind=link}
| HBV Protein | Molecules | ER Stress Signaling Pathway | Mode of Action | References |
|---|---|---|---|---|
| HBsAg | PERK | PERK-eIF2α pathway | Activator of GRP78 and PERK; HCC development and recurrence | Li et al., 2019 [66]; Su et al., 2008 [86] |
| IFN | UPR pathway | Inhibitor of UPR; Cell death promotion | Baudi et al., 2021 [87] | |
| HBx | IRE1α | IRE1α-XBP1 pathway | Activator of IRE1α; Increase EDEM; Apoptosis inhibition | Li et al., 2007 [63] |
| ATF6 | ATF6 pathway | Activator of ATF6 | Li et al., 2007 [63] | |
| SDF-1 | PERK-eIF2α pathway, ATF6 pathway | Activator of UPR; Cell survival promotion | Cho et al., 2011 [11]; Cho et al., 2014 [88] | |
| COX-2 | PERK-eIF2α-ATF4 pathway | Increase COX2 | Cho et al., 2011 [11] | |
| HBcAg | ATF6 | ATF6 pathway | Activate ATF6 and mediate cell apoptosis | Nakanishi et al.,2005 [89] |
| PERK | PERK-eIF2α pathway | Activator of GRP78 and PERK; | Choi et al., 2019 [90] | |
| IRE1α | IRE1α-XBP1 pathway | Activator of IRE1α; mediate cell apoptosis | Choi et al., 2019 [90] | |
| eIF2α | PERK-eIF2α pathway | Activator of eIF2α | Lee et al., 2015 [91] |
| HCV Protein | Molecules | ER Stress Signaling Pathway | Mode of Action | References |
|---|---|---|---|---|
| E2 | PERK | PERK-eIF2α pathway | Inhibitor of PERK | Zhang et al., 2012 [58] |
| Core | CHOP | ATF6 pathways | HCV core-mediated apoptosis | Takahashi et al., 2017 [127] Benali-Furet et al., 2005 [128] |
| NS2 | eIF2α | PERK-eIF2α pathway | eIF2α phosphorylation promotion | Bussche et al., 2010 [129] |
| NS3/4A | JNK | IRE1 pathway | Induce a mild apoptotic and oxidative stress response | Bussche et al., 2010 [129] Ríos-Ocampo et al., 2019 [130] |
| NS4B | XBP1, ATF6 | IRE1α-XBP1 pathway, ATF6 pathway | Activator of IRE1α and ATF6EDEM ↓ 1 | Zheng et al., 2005 [131] Tardif et al., 2004 [61] |
| NS5A | CHOP | PERK-eIF2α pathway | Increase the expression of CHOP | Meng et al., 2019 [132] Mishima et al., 2010 [133] Fang et al., 2013 [134] |
| E1 | MHC Ⅰ | ATF6 pathway | Activator of ATF6 | Tardif et al., 2003 [84] Selby et al.,1999 [135] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, T.; Wang, J.; Li, W.; Liu, M.; Han, N.; Yuan, M.; Du, L.; Tang, H. Endoplasmic Reticulum Stress in Hepatitis B Virus and Hepatitis C Virus Infection. Viruses 2022, 14, 2630. https://doi.org/10.3390/v14122630
Hu T, Wang J, Li W, Liu M, Han N, Yuan M, Du L, Tang H. Endoplasmic Reticulum Stress in Hepatitis B Virus and Hepatitis C Virus Infection. Viruses. 2022; 14(12):2630. https://doi.org/10.3390/v14122630
Chicago/Turabian StyleHu, Tengyue, Jiayi Wang, Weixiu Li, Miao Liu, Ning Han, Man Yuan, Lingyao Du, and Hong Tang. 2022. "Endoplasmic Reticulum Stress in Hepatitis B Virus and Hepatitis C Virus Infection" Viruses 14, no. 12: 2630. https://doi.org/10.3390/v14122630